Carnosine Prevents Aβ-Induced Oxidative Stress and Inflammation in Microglial Cells: A Key Role of TGF-β1

 ,
,  , , , ,
, , , ,
Abstract
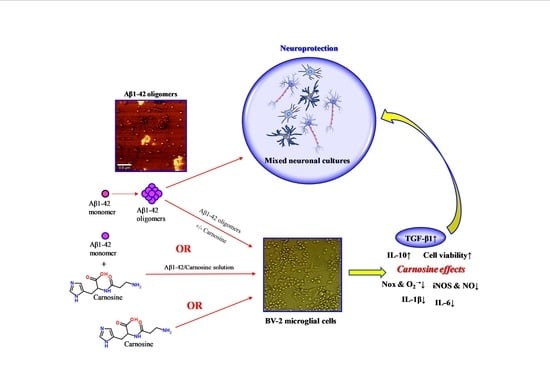
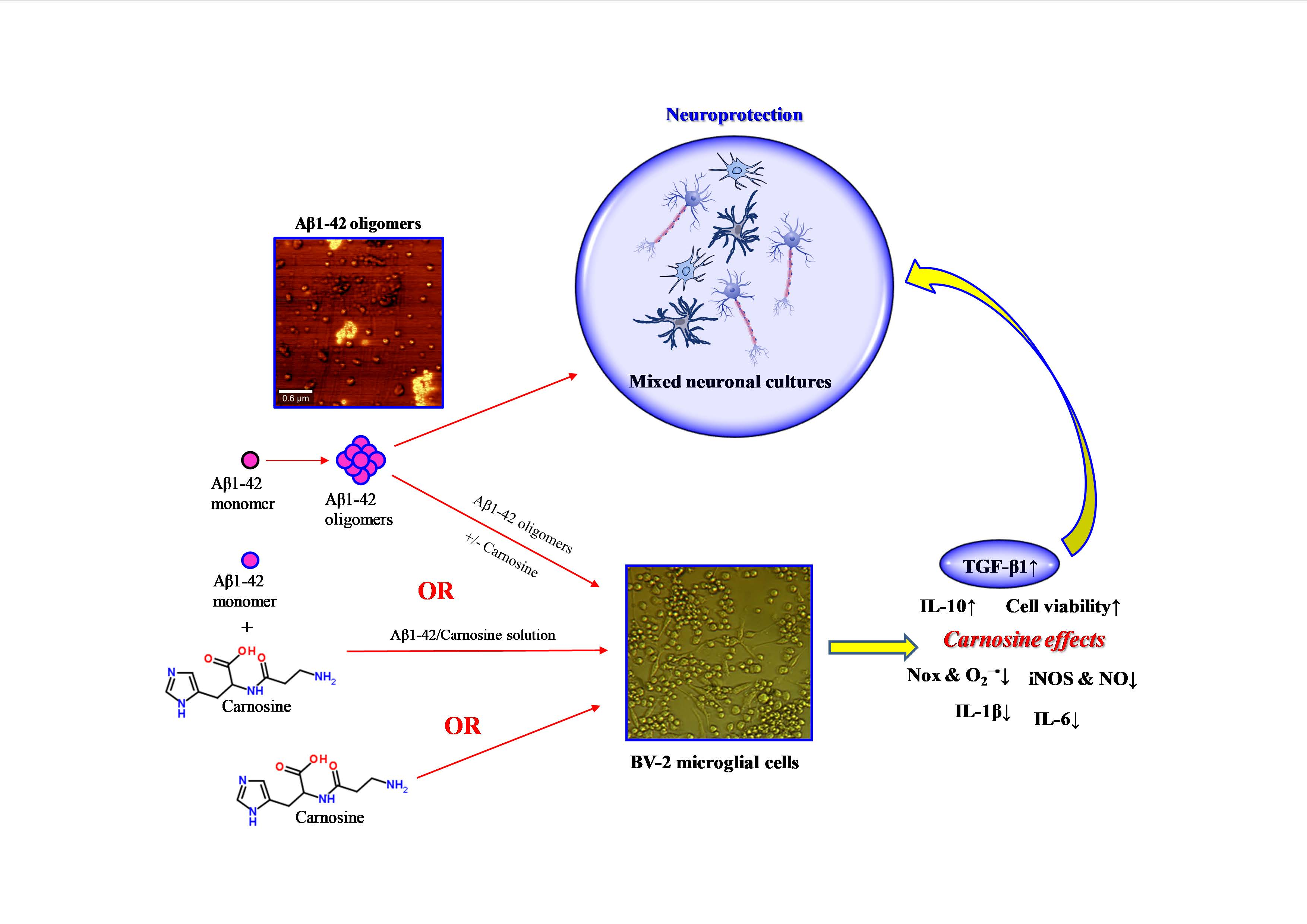
:
1. Introduction
2. Material and Methods
2.1. Materials and Reagents
2.2. Preparation of Aβ1-42 Oligomers
2.3. Atomic Force Microscope (AFM)
2.4. Cell Culture and Preparation
2.4.1. BV-2 Cells
2.4.2. Mixed Neuronal Cultures
2.5. Measurement of Cell Viability and Cell Death by the MTT and Trypan Blue Exclusion Assays
2.6. NO and O2−• Production Determination Using DAF-FM DA and MitoSOX Red Probes
2.7. Gene Expression Analysis by Quantitative Real-Time PCR (qRT-PCR)
2.8. Cytokine Secretion
2.9. Statistical Analysis
2.10. Study Approval
3. Results
3.1. Carnosine Protects BV-2 Cells Against Aβ1-42 Oligomers-Induced Cell Death
3.2. Carnosine Decreases Aβ1-42-Induced NO Production in Cultured Microglial Cells
3.3. Carnosine Decreases Aβ1-42-Induced O2−• Production in Cultured Microglial Cells
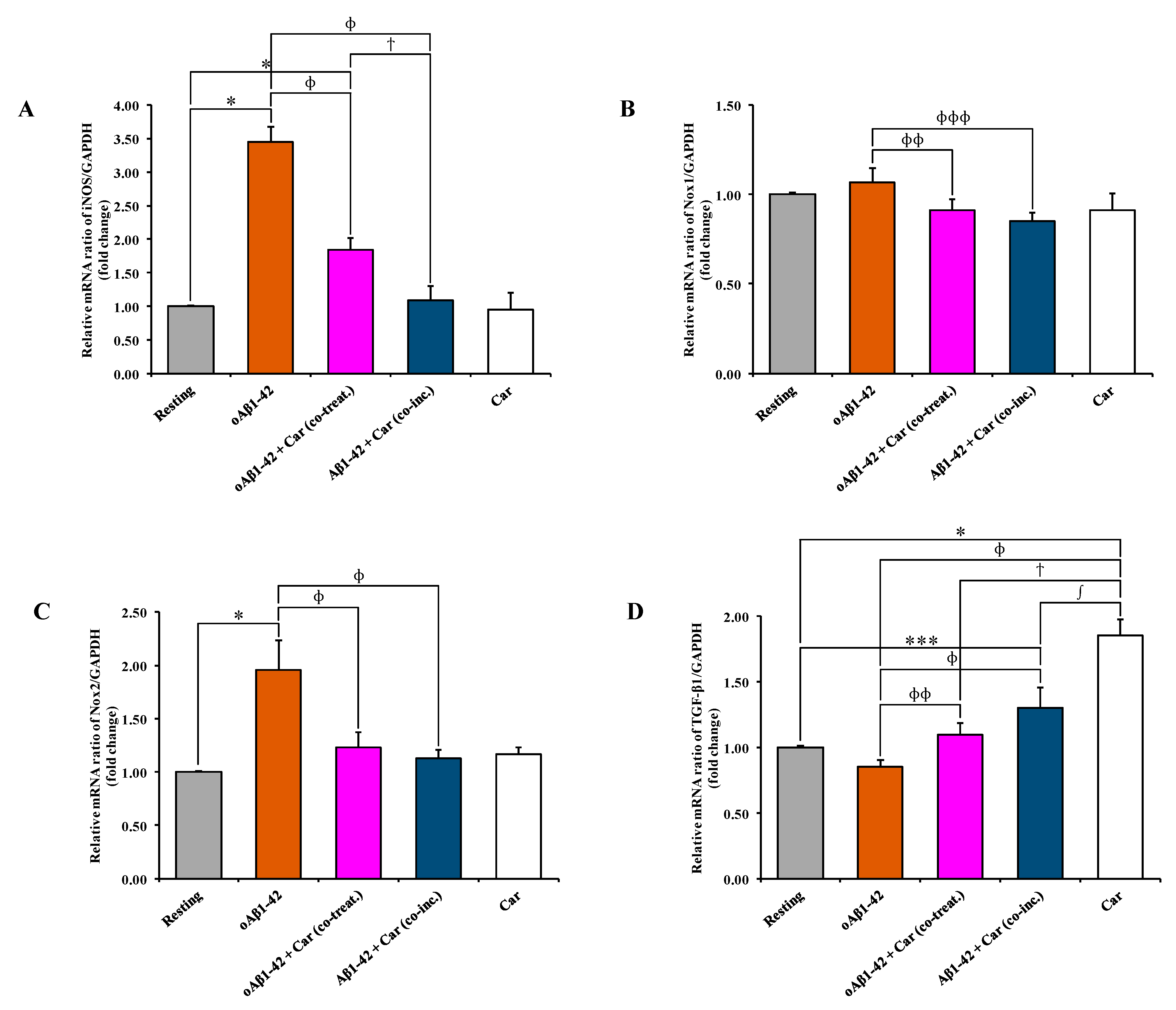
3.4. Carnosine Decreases Aβ1-42-Induced mRNA Expression Level of iNOS, Nox1, and Nox2 and Increases TGF-β1 mRNA Expression in Cultured Microglial Cells
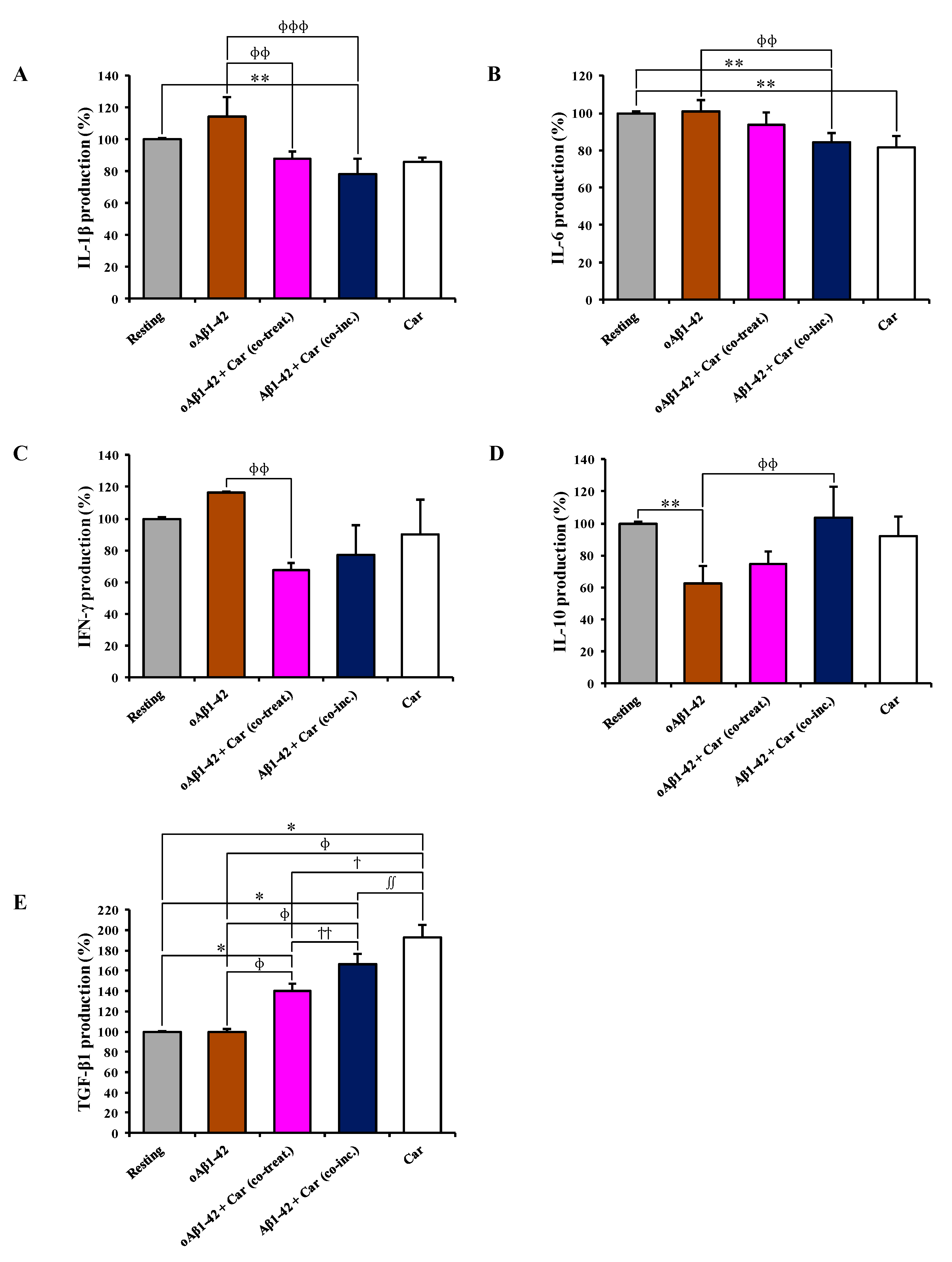
3.5. Carnosine Modulates the Release of Pro- and Anti-Inflammatory Cytokines in Cultured Microglial Cells
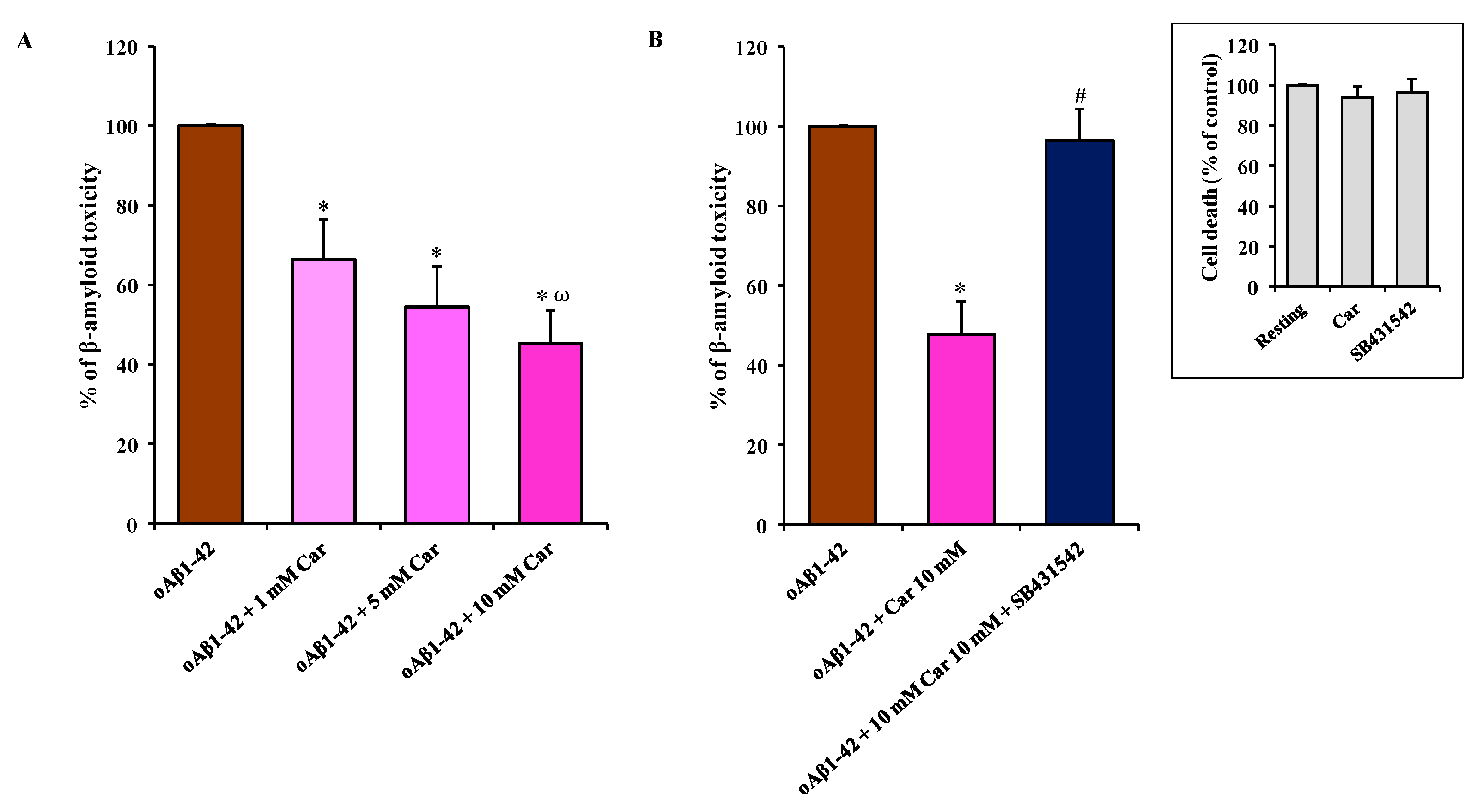
3.6. Carnosine Prevents oAβ1-42-Induced Toxicity in Mixed Neuronal Cultures via TGF-β1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Quinn, P.J.; Boldyrev, A.A.; Formazuyk, V.E. Carnosine: Its properties, functions and potential therapeutic applications. Mol. Asp. Med. 1992, 13, 379–444. [Google Scholar] [CrossRef]
- Hipkiss, A.R.; Brownson, C.; Bertani, M.F.; Ruiz, E.; Ferro, A. Reaction of carnosine with aged proteins: Another protective process? Ann. N. Y. Acad. Sci. 2002, 959, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Kulebyakin, K.; Karpova, L.; Lakonsteva, E.; Krasavin, M.; Boldyrev, A. Carnosine protects neurons against oxidative stress and modulates the time profile of mapk cascade signaling. Amino Acids 2012, 43, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Corona, C.; Frazzini, V.; Silvestri, E.; Lattanzio, R.; La Sorda, R.; Piantelli, M.; Canzoniero, L.M.; Ciavardelli, D.; Rizzarelli, E.; Sensi, S.L. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xtg-ad mice. PLoS ONE 2011, 6, e17971. [Google Scholar] [CrossRef]
- Aloisi, A.; Barca, A.; Romano, A.; Guerrieri, S.; Storelli, C.; Rinaldi, R.; Verri, T. Anti-aggregating effect of the naturally occurring dipeptide carnosine on abeta1-42 fibril formation. PLoS ONE 2013, 8, e68159. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Fresta, C.G.; Martinez-Becerra, F.; Antonio, L.; Johnson, R.T.; de Campos, R.P.S.; Siegel, J.M.; Wijesinghe, M.B.; Lazzarino, G.; Lunte, S.M. Carnosine modulates nitric oxide in stimulated murine raw 264.7 macrophages. Mol. Cell. Biochem. 2017, 431, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Guliaeva, N.V. [Superoxide-scavenging activity of carnosine in the presence of copper and zinc ions]. Biokhimiia 1987, 52, 1216–1220. [Google Scholar]
- Cripps, M.J.; Hanna, K.; Lavilla, C., Jr.; Sayers, S.R.; Caton, P.W.; Sims, C.; De Girolamo, L.; Sale, C.; Turner, M.D. Carnosine scavenging of glucolipotoxic free radicals enhances insulin secretion and glucose uptake. Sci. Rep. 2017, 7, 13313. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Younkin, S.G. Evidence that a beta 42 is the real culprit in alzheimer’s disease. Ann. Neurol. 1995, 37, 287–288. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Hung, A.Y.; Schlossmacher, M.G.; Oltersdorf, T.; Teplow, D.B.; Selkoe, D.J. Normal cellular processing of the beta-amyloid precursor protein results in the secretion of the amyloid beta peptide and related molecules. Ann. N. Y. Acad. Sci. 1993, 695, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P. Neurofibrillary tangles and alzheimer’s disease. Eur. Neurol. 1998, 40, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Brorsson, A.C.; Kumita, J.R.; MacLeod, I.; Bolognesi, B.; Speretta, E.; Luheshi, L.M.; Knowles, T.P.; Dobson, C.M.; Crowther, D.C. Methods and models in neurodegenerative and systemic protein aggregation diseases. Front. Biosci. 2010, 15, 373–396. [Google Scholar]
- Selkoe, D.J. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [PubMed]
- Kraft, A.D.; Harry, G.J. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int. J. Environ. Res. Public Health 2011, 8, 2980–3018. [Google Scholar] [CrossRef]
- Perlmutter, L.S.; Scott, S.A.; Barron, E.; Chui, H.C. Mhc class ii-positive microglia in human brain: Association with alzheimer lesions. J. Neurosci. Res. 1992, 33, 549–558. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- De Campos, R.P.; Siegel, J.M.; Fresta, C.G.; Caruso, G.; da Silva, J.A.; Lunte, S.M. Indirect detection of superoxide in raw 264.7 macrophage cells using microchip electrophoresis coupled to laser-induced fluorescence. Anal. Bioanal. Chem. 2015, 407, 7003–7012. [Google Scholar] [CrossRef]
- Mainz, E.R.; Gunasekara, D.B.; Caruso, G.; Jensen, D.T.; Hulvey, M.K.; da Silva, J.A.F.; Metto, E.C.; Culbertson, A.H.; Culbertson, C.T.; Lunte, S.M. Monitoring intracellular nitric oxide production using microchip electrophoresis and laser-induced fluorescence detection. Anal. Methods 2012, 4, 414–420. [Google Scholar] [CrossRef]
- Maes, M.; Galecki, P.; Chang, Y.S.; Berk, M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (NEURO)degenerative processes in that illness. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 676–692. [Google Scholar] [PubMed]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Preventing Ca2+-mediated nitrosative stress in neurodegenerative diseases: Possible pharmacological strategies. Cell Calcium 2010, 47, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Aktan, F. Inos-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Metto, E.C.; Evans, K.; Barney, P.; Culbertson, A.H.; Gunasekara, D.B.; Caruso, G.; Hulvey, M.K.; Fracassi da Silva, J.A.; Lunte, S.M.; Culbertson, C.T. An integrated microfluidic device for monitoring changes in nitric oxide production in single t-lymphocyte (jurkat) cells. Anal. Chem. 2013, 85, 10188–10195. [Google Scholar] [CrossRef]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef]
- Mander, P.; Brown, G.C. Activation of microglial nadph oxidase is synergistic with glial inos expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflamm. 2005, 2, 20. [Google Scholar] [CrossRef]
- Siegel, J.M.; Schilly, K.M.; Wijesinghe, M.B.; Caruso, G.; Fresta, C.G.; Lunte, S.M. Optimization of a microchip electrophoresis method with electrochemical detection for the determination of nitrite in macrophage cells as an indicator of nitric oxide production. Anal. Methods 2019, 11, 148–156. [Google Scholar] [CrossRef]
- Beckman, J.S.; Crow, J.P. Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochem. Soc. Trans. 1993, 21, 330–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, K.; Kohsaka, S. Microglia: Activation and their significance in the central nervous system. J. Biochem. 2001, 130, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Lucherini, O.M.; Lopalco, G.; Cantarini, L.; Emmi, G.; Lopalco, A.; Venerito, V.; Vitale, A.; Iannone, F. Critical regulation of Th17 cell differentiation by serum amyloid-A signalling in Behcet’s disease. Immunol Lett. 2018, 201, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Lopalco, G.; Lucherini, O.M.; Lopalco, A.; Venerito, V.; Fabiani, C.; Frediani, B.; Galeazzi, M.; Lapadula, G.; Cantarini, L.; Iannone, F. Cytokine Signatures in Mucocutaneous and Ocular Behçet’s Disease. Front. Immunol. 2017, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; O’Banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory processes in alzheimer’s disease. J. Neural Transm. 2010, 117, 919–947. [Google Scholar] [CrossRef] [PubMed]
- Rojo, L.E.; Fernandez, J.A.; Maccioni, A.A.; Jimenez, J.M.; Maccioni, R.B. Neuroinflammation: Implications for the pathogenesis and molecular diagnosis of alzheimer’s disease. Arch. Med. Res. 2008, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Schwab, C.; McGeer, P.L. Inflammatory aspects of alzheimer disease and other neurodegenerative disorders. J. Alzheimers Dis. 2008, 13, 359–369. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Beneventano, M.; Sortino, M.A. The contribution of microglia to early synaptic compensatory responses that precede β-amyloid-induced neuronal death. Sci. Rep. 2018, 8, 7297. [Google Scholar] [CrossRef]
- Caraci, F.; Busceti, C.; Biagioni, F.; Aronica, E.; Mastroiacovo, F.; Cappuccio, I.; Battaglia, G.; Bruno, V.; Caricasole, A.; Copani, A.; et al. The wnt antagonist, dickkopf-1, as a target for the treatment of neurodegenerative disorders. Neurochem. Res. 2008, 33, 2401–2406. [Google Scholar] [CrossRef]
- Caraci, F.; Spampinato, S.F.; Morgese, M.G.; Tascedda, F.; Salluzzo, M.G.; Giambirtone, M.C.; Caruso, G.; Munafo, A.; Torrisi, S.A.; Leggio, G.M.; et al. Neurobiological links between depression and ad: The role of tgf-beta1 signaling as a new pharmacological target. Pharmacol. Res. 2018, 130, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Luppi, C.; Fioravanti, M.; Bertolini, B.; Inguscio, M.; Grugnetti, A.; Guerriero, F.; Rovelli, C.; Cantoni, F.; Guagnano, P.; Marazzi, E.; et al. Growth factors decrease in subjects with mild to moderate alzheimer’s disease (AD): Potential correction with dehydroepiandrosterone-sulphate (DHEAS). Arch. Gerontol. Geriatr. 2009, 49 (Suppl. 1), 173–184. [Google Scholar] [CrossRef] [PubMed]
- Juraskova, B.; Andrys, C.; Holmerova, I.; Solichova, D.; Hrnciarikova, D.; Vankova, H.; Vasatko, T.; Krejsek, J. Transforming growth factor beta and soluble endoglin in the healthy senior and in alzheimer’s disease patients. J. Nutr. Health Aging 2010, 14, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Lin, C.; Yan, F.; Yu, G.Q.; Rohde, M.; McConlogue, L.; Masliah, E.; Mucke, L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med. 2001, 7, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Henn, A.; Lund, S.; Hedtjarn, M.; Schrattenholz, A.; Porzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 2009, 26, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Tascedda, F.; Merlo, S.; Benatti, C.; Spampinato, S.F.; Munafo, A.; Leggio, G.M.; Nicoletti, F.; Brunello, N.; Drago, F.; et al. Fluoxetine prevents abeta1-42-induced toxicity via a paracrine signaling mediated by transforming-growth-factor-beta1. Front. Pharmacol. 2016, 7, 389. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Molinaro, G.; Battaglia, G.; Giuffrida, M.L.; Riozzi, B.; Traficante, A.; Bruno, V.; Cannella, M.; Merlo, S.; Wang, X.; et al. Targeting group ii metabotropic glutamate (mglu) receptors for the treatment of psychosis associated with alzheimer’s disease: Selective activation of MGLU2 receptors amplifies beta-amyloid toxicity in cultured neurons, whereas dual activation of MGLU2 and MGLU3 receptors is neuroprotective. Mol. Pharmacol. 2011, 79, 618–626. [Google Scholar]
- Caruso, G.; Fresta, C.G.; Lazzarino, G.; Distefano, D.A.; Parlascino, P.; Lunte, S.M.; Lazzarino, G.; Caraci, F. Sub-toxic human amylin fragment concentrations promote the survival and proliferation of SH-SY5Y cells via the release of VEGF and HSPB5 from endothelial RBE4 cells. Int. J. Mol. Sci. 2018, 19, 3659. [Google Scholar] [CrossRef]
- Caruso, G.; Distefano, D.A.; Parlascino, P.; Fresta, C.G.; Lazzarino, G.; Lunte, S.M.; Nicoletti, V.G. Receptor-mediated toxicity of human amylin fragment aggregated by short- and long-term incubations with copper ions. Mol. Cell. Biochem. 2017, 425, 85–93. [Google Scholar] [CrossRef]
- Caruso, G.; Fresta, C.G.; Siegel, J.M.; Wijesinghe, M.B.; Lunte, S.M. Microchip electrophoresis with laser-induced fluorescence detection for the determination of the ratio of nitric oxide to superoxide production in macrophages during inflammation. Anal. Bioanal. Chem. 2017, 409, 4529–4538. [Google Scholar] [CrossRef]
- Gunasekara, D.B.; Siegel, J.M.; Caruso, G.; Hulvey, M.K.; Lunte, S.M. Microchip electrophoresis with amperometric detection method for profiling cellular nitrosative stress markers. Analyst 2014, 139, 3265–3273. [Google Scholar] [CrossRef] [PubMed]
- Fresta, C.G.; Chakraborty, A.; Wijesinghe, M.B.; Amorini, A.M.; Lazzarino, G.; Lazzarino, G.; Tavazzi, B.; Lunte, S.M.; Caraci, F.; Dhar, P.; et al. Non-toxic engineered carbon nanodiamond concentrations induce oxidative/nitrosative stress, imbalance of energy metabolism, and mitochondrial dysfunction in microglial and alveolar basal epithelial cells. Cell Death Dis. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Barresi, V.; Ragusa, A.; Fichera, M.; Musso, N.; Castiglia, L.; Rappazzo, G.; Travali, S.; Mattina, T.; Romano, C.; Cocchi, G.; et al. Decreased expression of GRAF1/ophn-1-l in the x-linked alpha thalassemia mental retardation syndrome. BMC Med. Genom. 2010, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Pappalardo, G.; Basile, L.; Giuffrida, A.; Copani, A.; Tosto, R.; Sinopoli, A.; Giuffrida, M.L.; Pirrone, E.; Drago, F.; et al. Neuroprotective effects of the monoamine oxidase inhibitor tranylcypromine and its amide derivatives against abeta(1-42)-induced toxicity. Eur. J. Pharmacol. 2015, 764, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.L. Synaptotoxic amyloid-beta oligomers: A molecular basis for the cause, diagnosis, and treatment of alzheimer’s disease? J. Alzheimers Dis. 2013, 33 (Suppl. 1), S49–S65. [Google Scholar] [CrossRef] [PubMed]
- Jiao, C.; Gao, F.; Ou, L.; Yu, J.; Li, M.; Wei, P.; Miu, F. Tetrahydroxystilbene glycoside antagonizes beta-amyloid-induced inflammatory injury in microglia cells by regulating pu.1 expression. Neuroreport 2018, 29, 787–793. [Google Scholar] [CrossRef]
- Ries, M.; Loiola, R.; Shah, U.N.; Gentleman, S.M.; Solito, E.; Sastre, M. The anti-inflammatory annexin a1 induces the clearance and degradation of the amyloid-beta peptide. J. Neuroinflamm. 2016, 13, 234. [Google Scholar] [CrossRef]
- Butterfield, D.A. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Serini, S.; Calviello, G. Reduction of oxidative/nitrosative stress in brain and its involvement in the neuroprotective effect of n-3 pufa in alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Gelain, D.P.; Antonio Behr, G.; Birnfeld de Oliveira, R.; Trujillo, M. Antioxidant therapies for neurodegenerative diseases: Mechanisms, current trends, and perspectives. Oxid. Med. Cell. Longev. 2012, 2012, 895153. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol. 2000, 130, 184–208. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [PubMed]
- Togo, T.; Katsuse, O.; Iseki, E. Nitric oxide pathways in alzheimer’s disease and other neurodegenerative dementias. Neurol. Res. 2004, 26, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Pautler, R.G.; Klann, E. Mitochondrial superoxide: A key player in alzheimer’s disease. Aging 2009, 1, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef] [Green Version]
- Seubert, P.; Vigo-Pelfrey, C.; Esch, F.; Lee, M.; Dovey, H.; Davis, D.; Sinha, S.; Schlossmacher, M.; Whaley, J.; Swindlehurst, C.; et al. Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature 1992, 359, 325–327. [Google Scholar] [CrossRef]
- Strozyk, D.; Blennow, K.; White, L.R.; Launer, L.J. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 2003, 60, 652–656. [Google Scholar] [CrossRef]
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury, P.T. Jr. Assembly of A beta amyloid protofibrils: An in vitro model for a possible early event in Alzheimer’s disease. Biochemistry 1999, 38, 8972–8980. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, A.; Teplow, D.B.; Kirschner, D.A.; Benedek, G.B. Kinetic theory of fibrillogenesis of amyloid beta-protein. Proc. Natl. Acad. Sci. USA 1997, 94, 7942–7947. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-beta peptides by microglia and macrophages: The issue of what, when and where. Future Neurol. 2012, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Fleisher-Berkovich, S.; Abramovitch-Dahan, C.; Ben-Shabat, S.; Apte, R.; Beit-Yannai, E. Inhibitory effect of carnosine and n-acetyl carnosine on lps-induced microglial oxidative stress and inflammation. Peptides 2009, 30, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Lopachev, A.V.; Lopacheva, O.M.; Abaimov, D.A.; Koroleva, O.V.; Vladychenskaya, E.A.; Erukhimovich, A.A.; Fedorova, T.N. Neuroprotective effect of carnosine on primary culture of rat cerebellar cells under oxidative stress. Biochemistry 2016, 81, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Klebanov, G.I.; Teselkin Yu, O.; Babenkova, I.V.; Popov, I.N.; Levin, G.; Tyulina, O.V.; Boldyrev, A.A.; Vladimirov Yu, A. Evidence for a direct interaction of superoxide anion radical with carnosine. Biochem. Mol. Biol. Int. 1997, 43, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresta, C.G.; Hogard, M.L.; Caruso, G.; Melo Costa, E.E.; Lazzarino, G.; Lunte, S.M. Monitoring carnosine uptake by raw 264.7 macrophage cells using microchip electrophoresis with fluorescence detection. Anal. Methods 2017, 9, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, F.; Cataldo, S.; Fisichella, S.; Nicoletti, S.; Nicoletti, V.G.; Pignataro, B.; Savarino, A.; Rizzarelli, E. Protective effects of l- and d-carnosine on alpha-crystallin amyloid fibril formation: Implications for cataract disease. Biochemistry 2009, 48, 6522–6531. [Google Scholar] [CrossRef]
- Javadi, S.; Yousefi, R.; Hosseinkhani, S.; Tamaddon, A.M.; Uversky, V.N. Protective effects of carnosine on dehydroascorbate-induced structural alteration and opacity of lens crystallins: Important implications of carnosine pleiotropic functions to combat cataractogenesis. J. Biomol. Struct. Dyn. 2017, 35, 1766–1784. [Google Scholar] [CrossRef]
- Salim-Hanna, M.; Lissi, E.; Videla, L.A. Free radical scavenging activity of carnosine. Free Radic. Res. Commun. 1991, 14, 263–270. [Google Scholar] [CrossRef]
- Hipkiss, A.R.; Michaelis, J.; Syrris, P. Non-enzymatic glycosylation of the dipeptide l-carnosine, a potential anti-protein-cross-linking agent. FEBS Lett. 1995, 371, 81–85. [Google Scholar] [CrossRef]
- Grasso, G.I.; Bellia, F.; Arena, G.; Satriano, C.; Vecchio, G.; Rizzarelli, E. Multitarget trehalose-carnosine conjugates inhibit abeta aggregation, tune copper(ii) activity and decrease acrolein toxicity. Eur. J. Med. Chem. 2017, 135, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, F.; Convertino, M.; Magno, A.; Caflisch, A.; Corazza, A.; Haridas, H.; Esposito, G.; Cataldo, S.; Pignataro, B.; Milardi, D.; et al. Carnosine inhibits abeta(42) aggregation by perturbing the h-bond network in and around the central hydrophobic cluster. Chembiochem 2013, 14, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, M.L.; Caraci, F.; De Bona, P.; Pappalardo, G.; Nicoletti, F.; Rizzarelli, E.; Copani, A. The monomer state of beta-amyloid: Where the alzheimer’s disease protein meets physiology. Rev. Neurosci. 2010, 21, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, M.L.; Tomasello, M.F.; Pandini, G.; Caraci, F.; Battaglia, G.; Busceti, C.; Di Pietro, P.; Pappalardo, G.; Attanasio, F.; Chiechio, S.; et al. Monomeric ss-amyloid interacts with type-1 insulin-like growth factor receptors to provide energy supply to neurons. Front. Cell. Neurosci. 2015, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- Hashioka, S.; Miyaoka, T.; Wake, R.; Furuya, M.; Horiguchi, J. Glia: An important target for anti-inflammatory and antidepressant activity. Curr. Drug Targets 2013, 14, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, D.; Mizrahi, R. Molecular imaging of neuroinflammation in alzheimer’s disease and mild cognitive impairment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 123–131. [Google Scholar] [CrossRef]
- Businaro, R.; Corsi, M.; Asprino, R.; Di Lorenzo, C.; Laskin, D.; Corbo, R.M.; Ricci, S.; Pinto, A. Modulation of inflammation as a way of delaying alzheimer’s disease progression: The diet’s role. Curr. Alzheimer Res. 2018, 15, 363–380. [Google Scholar] [CrossRef]
- Pangestuti, R.; Vo, T.S.; Ngo, D.H.; Kim, S.K. Fucoxanthin ameliorates inflammation and oxidative reponses in microglia. J. Agric. Food Chem. 2013, 61, 3876–3883. [Google Scholar] [CrossRef]
- Correani, V.; Di Francesco, L.; Cera, I.; Mignogna, G.; Giorgi, A.; Mazzanti, M.; Fumagalli, L.; Fabrizi, C.; Maras, B.; Schinina, M.E. Reversible redox modifications in the microglial proteome challenged by beta amyloid. Mol. Biosyst. 2015, 11, 1584–1593. [Google Scholar] [CrossRef]
- Hatakeyama, D.; Kozawa, O.; Otsuka, T.; Shibata, T.; Uematsu, T. Zinc suppresses IL-6 synthesis by prostaglandin F2alpha in osteoblasts: Inhibition of phospholipase C and phospholipase D. J. Cell. Biochem. 2002, 85, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yan, H.; Wang, R.; Bo, S.; Lu, X.; Zhang, J.; Xu, A. Protective effect of carnosine on white matter damage in corpus striatum induced by chronic cerebral hypoperfusion. Neurosci. Lett. 2018, 683, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.X.; Li, D.W.; Liu, X.C.; Yang, M.F.; Fang, J.; Sun, B.L.; Zhang, Z.Y.; Yang, X.Y. Carnosine attenuates brain oxidative stress and apoptosis after intracerebral hemorrhage in rats. Neurochem. Res. 2017, 42, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Sun, B.L.; Yang, M.F.; Li, D.W.; Fang, J.; Zhang, S. Carnosine attenuates early brain injury through its antioxidative and anti-apoptotic effects in a rat experimental subarachnoid hemorrhage model. Cell. Mol. Neurobiol. 2015, 35, 147–157. [Google Scholar] [CrossRef]
- Ma, J.; Xiong, J.Y.; Hou, W.W.; Yan, H.J.; Sun, Y.; Huang, S.W.; Jin, L.; Wang, Y.; Hu, W.W.; Chen, Z. Protective effect of carnosine on subcortical ischemic vascular dementia in mice. CNS Neurosci. Ther. 2012, 18, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, C.A.; Carvalho, M.D.G.; Sousa, L.P.; Caramelli, P.; Gomes, K.B. Alzheimer’s disease and cytokine il-10 gene polymorphisms: Is there an association? Arq Neuropsiquiatr. 2017, 75, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Ke, K.F.; Lu, J.H.; Qiu, Y.H.; Peng, Y.P. Protection of tgf-beta1 against neuroinflammation and neurodegeneration in abeta1-42-induced alzheimer’s disease model rats. PLoS ONE 2015, 10, e0116549. [Google Scholar]
- Brionne, T.C.; Tesseur, I.; Masliah, E.; Wyss-Coray, T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 2003, 40, 1133–1145. [Google Scholar] [CrossRef]
- Tichauer, J.E.; Flores, B.; Soler, B.; Eugenin-von Bernhardi, L.; Ramirez, G.; von Bernhardi, R. Age-dependent changes on TGFBETA1 SMAD3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 2014, 37, 187–196. [Google Scholar] [CrossRef]
- Affram, K.O.; Mitchell, K.; Symes, A.J. Microglial activation results in inhibition of tgf-beta-regulated gene expression. J. Mol. Neurosci. 2017, 63, 308–319. [Google Scholar] [CrossRef]
- Caruso, G.; Caraci, F.; Jolivet, R.B. Pivotal role of carnosine in the modulation of brain cells activity: Multimodal mechanism of action and therapeutic potential in neurodegenerative disorders. Prog. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tesseur, I.; Zou, K.; Esposito, L.; Bard, F.; Berber, E.; Can, J.V.; Lin, A.H.; Crews, L.; Tremblay, P.; Mathews, P.; et al. Deficiency in neuronal tgf-beta signaling promotes neurodegeneration and alzheimer’s pathology. J. Clin. Investig. 2006, 116, 3060–3069. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Gili, E.; Calafiore, M.; Failla, M.; La Rosa, C.; Crimi, N.; Sortino, M.A.; Nicoletti, F.; Copani, A.; Vancheri, C. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol. Res. 2008, 57, 274–282. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
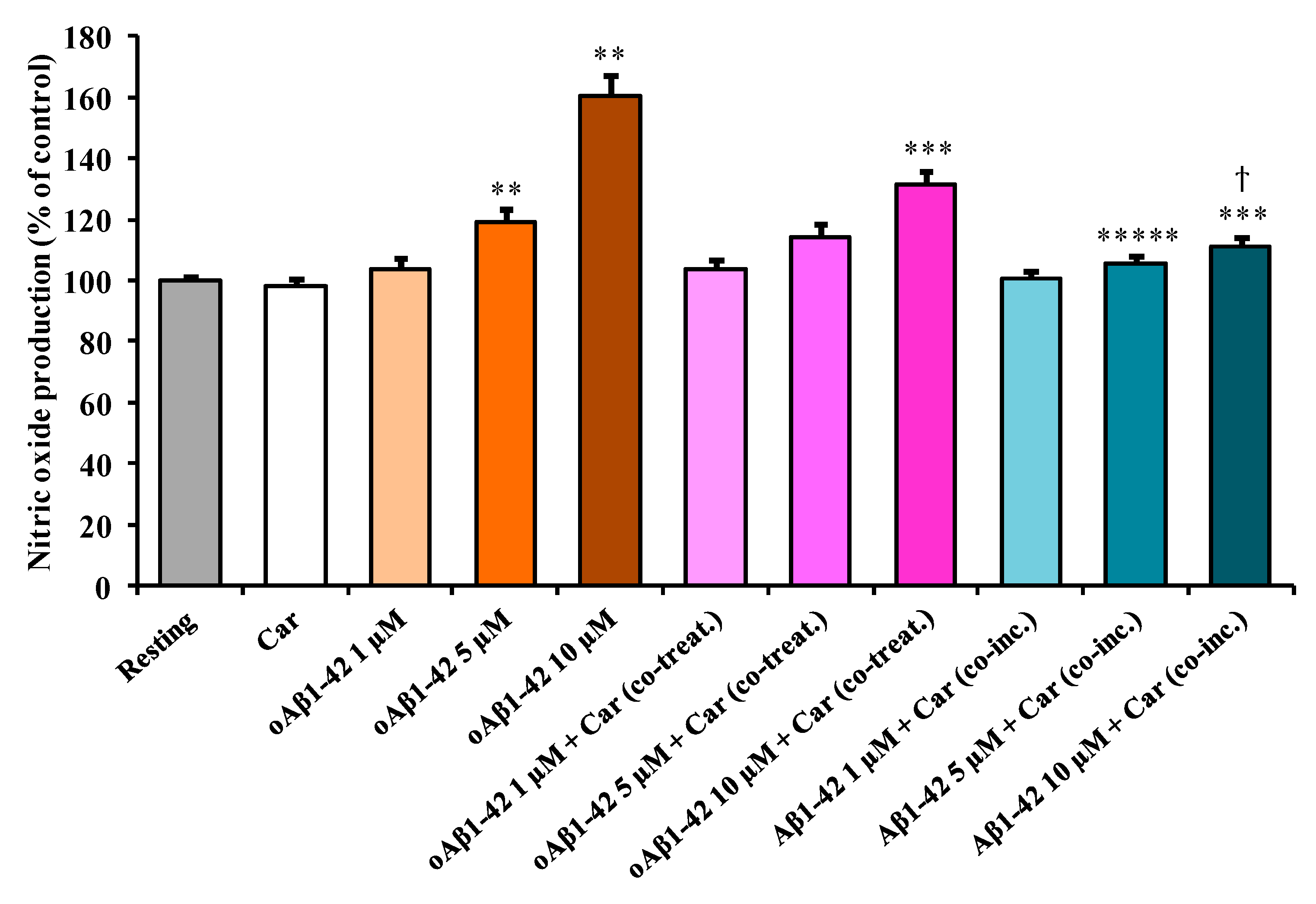{kind=link}
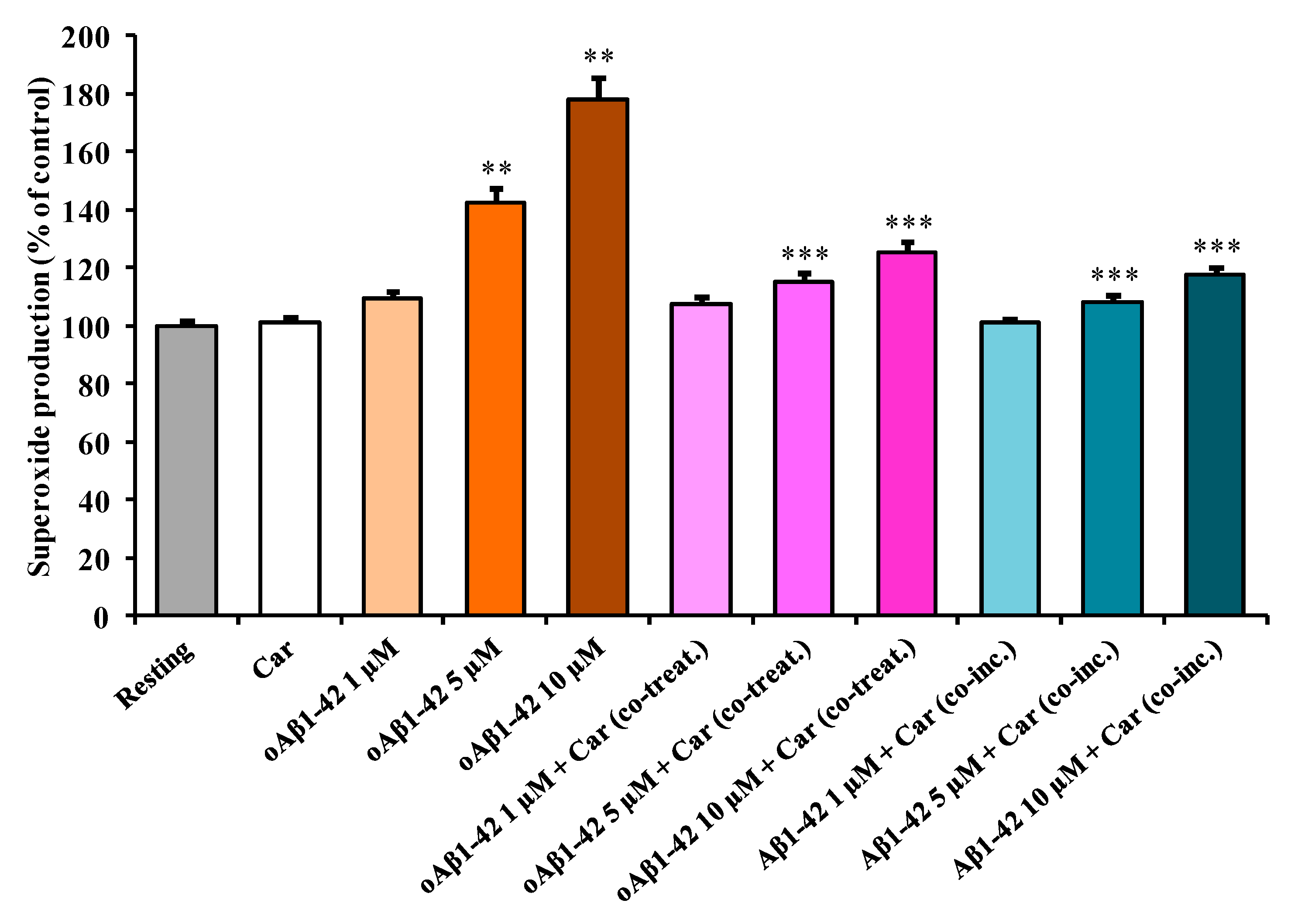{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Official Name # | Official Symbol | Alternative Titles/Symbols | Detected Transcript | Amplicon Length | Cat. No. § |
|---|---|---|---|---|---|
| nitric oxide synthase 2, inducible | Nos2 | iNOS; Nos-2; Nos2a; i-NOS; NOS-II; MAC-NOS | NM_010927 | 118 bp | QT00100275 |
| NADPH oxidase 1 | Nox1 | MOX1; NOH1; NOH-1; NOX1a; Nox-1; GP91-2; NOX1alpha | NM_172203 XM_006528515 | 180 bp | QT00140091 |
| cytochrome b-245, beta polypeptide | Cybb | Cgd; Cyd; Nox2; C88302; gp91-1; gp91phox; CGD91-phox | NM_007807 XM_006527565 | 146 bp | QT00139797 |
| transforming growth factor, beta 1 | Tgfb1 | Tgfb; Tgfb-1; TGFbeta1; TGF-beta1 | NM_011577 | 145 bp | QT00145250 |
| interleukin 6 | Il6 | Il-6 | NM_031168 | 128 bp | QT00098875 |
| glyceraldehyde-3-phosphate dehydrogenase | Gapdh | Gapd | NM_008084 XM_001003314 XM_990238 NM_001289726 | 144 bp | QT01658692 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caruso, G.; Fresta, C.G.; Musso, N.; Giambirtone, M.; Grasso, M.; Spampinato, S.F.; Merlo, S.; Drago, F.; Lazzarino, G.; Sortino, M.A.; et al. Carnosine Prevents Aβ-Induced Oxidative Stress and Inflammation in Microglial Cells: A Key Role of TGF-β1. Cells 2019, 8, 64. https://doi.org/10.3390/cells8010064
Caruso G, Fresta CG, Musso N, Giambirtone M, Grasso M, Spampinato SF, Merlo S, Drago F, Lazzarino G, Sortino MA, et al. Carnosine Prevents Aβ-Induced Oxidative Stress and Inflammation in Microglial Cells: A Key Role of TGF-β1. Cells. 2019; 8(1):64. https://doi.org/10.3390/cells8010064
Chicago/Turabian StyleCaruso, Giuseppe, Claudia G. Fresta, Nicolò Musso, Mariaconcetta Giambirtone, Margherita Grasso, Simona F. Spampinato, Sara Merlo, Filippo Drago, Giuseppe Lazzarino, Maria A. Sortino, and et al. 2019. "Carnosine Prevents Aβ-Induced Oxidative Stress and Inflammation in Microglial Cells: A Key Role of TGF-β1" Cells 8, no. 1: 64. https://doi.org/10.3390/cells8010064