Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease


1
Department of Microbiology, Chungnam National University School of Medicine, Daejeon 35015, Korea
2
Department of Medical Science, Chungnam National University School of Medicine, Daejeon 35015, Korea
3
Infection Control Convergence Research Center, Chungnam National University School of Medicine, Daejeon 35015, Korea
4
Department of Radiation Oncology, Chungnam National University Hospital, 282, Munwha-ro, Jung-gu, Daejeon 34952, Korea
5
Department of Internal Medicine, Chungnam National University Hospital, 282, Munwha-ro, Jung-gu, Daejeon 34952, Korea
6
Department of Internal Medicine, School of Medicine, Chungnam National University, 266, Munwha-ro, Jung-gu, Daejeon 35015, Korea
*
Author to whom correspondence should be addressed.
Cells 2019, 8(1), 77; https://doi.org/10.3390/cells8010077
Submission received: 15 December 2018
/
Revised: 9 January 2019
/
Accepted: 11 January 2019
/
Published: 21 January 2019
(This article belongs to the Special Issue Autophagy in Tissue Injury and Homeostasis)
Abstract
:Autophagy is an intracellular catabolic process that is essential for a variety of cellular responses. Due to its role in the maintenance of biological homeostasis in conditions of stress, dysregulation or disruption of autophagy may be linked to human diseases such as inflammatory bowel disease (IBD). IBD is a complicated inflammatory colitis disorder; Crohn’s disease and ulcerative colitis are the principal types. Genetic studies have shown the clinical relevance of several autophagy-related genes (ATGs) in the pathogenesis of IBD. Additionally, recent studies using conditional knockout mice have led to a comprehensive understanding of ATGs that affect intestinal inflammation, Paneth cell abnormality and enteric pathogenic infection during colitis. In this review, we discuss the various ATGs involved in macroautophagy and selective autophagy, including ATG16L1, IRGM, LRRK2, ATG7, p62, optineurin and TFEB in the maintenance of intestinal homeostasis. Although advances have been made regarding the involvement of ATGs in maintaining intestinal homeostasis, determining the precise contribution of autophagy has remained elusive. Recent efforts based on direct targeting of ATGs and autophagy will further facilitate the development of new therapeutic opportunities for IBD.
1. Introduction
Inflammatory bowel disease (IBD) is a complicated autoimmune disorder with multiple etiologies including genetic predisposition, environmental factors and immune-associated pathogenesis [1]. Both Crohn’s disease (CD) and ulcerative colitis (UC), the major clinical phenotypes of IBD, are systemic diseases associated with autoimmune manifestations [2,3]. Although the intestinal host defense is maintained by balancing inflammation and the immune response, excessive inflammation may damage the intestine and its mucosal barrier [1,3]. Although IBD is known to be a polygenic disorder, emerging evidence indicates that genetic susceptibility associated with host autophagy is an important factor in the pathogenesis of IBD [4,5].
Autophagy is a cytosolic process that triggers lysosomal degradation of cytosolic materials to maintain intracellular homeostasis under conditions of stress by recycling metabolic building blocks [6,7]. Intracellular cargos sequestered by autophagosomes include damaged cellular organelles, large protein aggregates and intracellular pathogens [8,9]. It is now clear that activation of autophagy contributes to the amelioration of excessive inflammatory responses [10,11]. Dysfunctional or dysregulated autophagy can lead to diverse inflammatory, immune and metabolic disorders [10,11]. Previous studies have demonstrated the involvement of genetic variations of autophagy genes, including ATG16L1 and IRGM, in the pathogenesis of colitis [12,13,14,15,16,17,18]. More recently, it was shown that autophagy-related genes (ATGs), such as optineurin (OPTN), transcription factor EB (TFEB) and leucine-rich repeat kinase (LRRK), are associated with increased susceptibility to colitis, suggesting that these genes are important in colonic immune homeostasis [19,20,21,22,23,24,25].
This review will focus on recent progress in elucidating the roles of ATGs in colonic inflammation and their clinical relevance. We will highlight recent findings regarding several ATGs and the mechanisms through which colitis severity is regulated.
2. Overview of Autophagy, Selective Autophagy and ATGs
Macroautophagy (herein referred to as autophagy) is an intracellular catabolic process through which cytoplasmic cargos are sequestered and delivered to lysosomes for degradation [26]. Autophagy plays a critical role in the maintenance of cellular homeostasis during a variety of stress responses, including starvation, hypoxia, toxicity and inflammation [27]. Although it was originally believed that autophagy was a nonspecific process that occurred under starvation conditions, it is now known that autophagy can target specific intracellular organelles or foreign pathogens for timely degradation, which is known as selective autophagy. A detailed discussion of the general aspects of autophagy is beyond the scope of this review; there are numerous excellent reviews dealing in detail about autophagy [28,29]. Here, we briefly review nonselective and selective autophagy, as well as ATGs (Figure 1), before focusing on the relationship of ATGs and autophagy with the pathogenesis of IBD.
2.1. Autophagy
Autophagy plays an important housekeeping function in cells through the removal of superfluous or damaged organelles in lysosomes. The autophagic process consists of multiple stages: initiation and biogenesis of autophagosomes, followed by maturation and fusion with lysosomes (Figure 1A) [30]. Autophagy is initiated by the formation of the phagophore, in which the edges of isolation membranes elongate and engulf cytoplasmic cargos [30]. Once the double-membrane structure contains cytoplasmic cargos, autophagosomes undergo maturation to form a completed autophagosome structure and ultimately are fused with a late endosome and lysosome to initiate degradation of cargos [31,32].
Regulation of autophagy is important to prevent cell death and pathogenic conditions [33]. It is now clear that autophagic activity is tightly regulated by molecular machinery and transcription factors at transcriptional and post-translational levels [33]. Recent studies have identified several nuclear transcription factors that coordinate autophagy via transcriptional activation of ATGs [33]. For example, TFEB plays an essential role in lysosomal biogenesis and activity during autophagy (Figure 2) [34]. We also briefly highlight the involvement of TFEB in the regulation of colonic inflammation (Figure 2).
2.2. Selective Autophagy
In addition to nonselective degradation, autophagy also plays a role in the targeting and clearance of specific targets/substrates, that is, selective autophagy, which is named depending on its specific targets and includes mitophagy, xenophagy and aggrephagy (Figure 1B) [35,36,37,38]. Selective autophagy involves several steps, including a degradation cue, cargo recognition via selective autophagy receptors, ubiquitination and the recruitment of autophagosome machinery but it does not necessarily occur in a stepwise manner [39,40]. Several ubiquitin binding proteins, such as p62, neighbor of breast cancer 1 (NBR1), OPTN and NDP52/CALCOCO2, have been identified as autophagy receptors responsible for the delivery of ubiquitinated cargos to the autophagy system [41,42,43,44,45,46]. Cargo signals can be classified as ubiquitin-dependent and -independent recognition [40]. Autophagic cargo receptors contain the LC3-interacting region (LIR) motif, which connects cargos to ATG8 family proteins for selective autophagic degradation [45,46]. Despite advances in knowledge of the mechanisms and players involved in canonical and noncanonical autophagy, we still lack a clear understanding of its function in a variety of physiologic and pathologic responses. A few reports have demonstrated the involvement of several autophagic receptors, including p62 and OPTN, in the control of intestinal homeostasis. However, additional autophagic receptors or regulators of autophagic signaling pathways must operate to ameliorate excessive colonic inflammation. Finally, it will be important to investigate the mechanisms by which autophagic receptors or ATGs impact clinical outcomes.
2.3. ATGs and the Control of Autophagy
Each step of the autophagy process is highly orchestrated by numerous ATGs; nearly 40 ATGs have been identified in yeast and orthologs of yeast ATGs have been identified in higher eukaryotes with some exceptions, such as mammalian ATG101 [47,48,49]. Among these ATGs, certain gene groups are required for autophagosome formation and are shared among various types of autophagy, such as nonselective and selective autophagy. Mammalian ATGs can be divided into several functional clusters including the ULK1-ATG13-FIP200-ATG101 protein kinase complex, the PtdIns3K class III complex containing VPS34, VPS15 and Beclin 1, the ubiquitin-like ATG5/ATG12 complex and the ubiquitin-like ATG8/LC3 conjugation system [50]. These ATGs participate in different stages of autophagy, such as the induction of autophagosome formation, expansion of phagophores and autophagosome completion [50,51].
Recent studies have identified and reported the roles of numerous cargo receptors including NBR1, multi-domain scaffold/adaptor protein p62/sequestosome-1 (p62/SQSTM-1), nuclear domain 10 protein 52 (NDP52) and OPTN [41,42,44]. These selective autophagic receptors contain the LIR motif, thereby connecting ubiquitin-tagged substrates to ATG8 family members such as microtubule-associated protein 1A/1B-light chain 3/γ-aminobutyric acid receptor-associated protein (LC3/GABARAP) [42,45,52].
3. Overview of IBD
IBD is a disease in which chronic inflammation of intestinal cells occurs due to unknown causes [53]. CD and UC are classified according to the clinical features and characteristics of the disease. Both diseases have similar clinical symptoms including diarrhea, abdominal pain, hematochezia and weight loss; however, the location of inflammation, infiltration degree and complications differ [54]. In general, CD is known to mediate Th1 cell-mediated inflammatory responses and UC is known to mediate Th2 cells [55]. Recently, loss of the suppressive functions of interleukin (IL)-17A-producing regulatory T cells was reported to cause IBD [56]. In general, both types of IBD are treated with anti-inflammatory drugs, such as 5-aminosalicylic acid and corticosteroids; however, in the absence of clinical improvement following treatment with anti-inflammatory drugs, patients achieved a high remission rate using anti-tumor necrosis factor (TNF)-α drugs [57]. However, more than one-third of IBD patients do not respond to anti-TNF-α drugs [58]. Recently, a new therapeutic target for IBD has emerged and the role of Paneth cells in intestinal homeostasis is discussed.
Crypts, concave structures of granulated cells clustered in the base of the small intestine, contain 5–12 Paneth cells. Unlike ordinary enterocytes, which have an average lifespan of 3–5 days, Paneth cells have a longer life expectancy of 20 days [59]. Paneth cells can differentiate into three different cell lineages: enterocytes, goblet cells and enteroendocrine cells [60]. Paneth cells exhibit antimicrobial effects by secreting secretory granules containing antimicrobial peptides (AMPs) and other peptides including lysozymes, alpha-defensins and secretory phospholipase A1 in response to cell stimuli to crypt lumen [61,62,63]. Paneth cell secretion of AMPs plays an important role not only in clearing invading pathogens but also in maintaining the diversity and quantity of intestinal microbiota via intestinal antimicrobial function [64]. Although Paneth cells are normally localized to the small intestine, diseases such as chronic inflammation may result in intestinal metaplasia, which is characterized by the localization and function of Paneth cells in aberrant sites, such as the colon [65]. These metaplastic Paneth cells protect the colonic epithelium from bacterial invasion [59]. However, Paneth cell loss may occur in situations with acute inflammation such as Grade II/III graft versus host disease or in CD [66,67]. In this case, Paneth cells are replaced with lysozyme-producing mucus cells, which can be followed by the development of diseases such as IBD in the small intestine [68].
Paneth cells present in the intestinal epithelium are essential for maintaining the homeostasis of normal colonizing microbes of the host. The important pathway in this process is xenophagy, an autophagic pathogen removal process that allows the host to maintain normal metabolic function [69,70]. However, when dysfunction of Paneth cells occurs due to environmental or genetic influences, AMPs are not secreted properly. As a result, dysbiosis, a discrepancy in the composition of normal intestinal microbiota, occurs, which is an important cause of intestinal disorders, especially IBD [71]. For example, in CD patients with impaired xenophagy, adherent-invasive Escherichia coli (AIEC) or Salmonella typhimurium colonize intestinal epithelial cells (IECs) due to the autophagic dysfunction of Paneth cells [72,73,74]. Thus, the impairment of autophagy in Paneth cells makes it difficult to treat incoming pathogenic bacteria as well as to respond to alterations in the composition of the intestinal microbiota [75]. Ultimately, the poor xenophagy of Paneth cells makes the intestinal epithelium hypersensitive to infiltrating microbes or their products and promotes bacterial self-proliferation and the onset of IBD [76,77].
4. ATG Involvement in IBD Pathogenesis
The clinical diversity and heterogeneity of the IBD phenotype are likely due to the presence of genetic heterogeneity together with environmental factors. Susceptibility to IBD may be due to an interaction of several genes, identified by genome-wide association studies (GWASs) [78,79,80]. To date, over 200 loci have been identified as genetically significant loci by a meta-analysis combined with GWAS [81,82]. Earlier independent GWASs showed that autophagy gene variants, including autophagy-related gene 16-like 1 (ATG16L1) and immunity-related GTPase M (IRGM) are linked to CD susceptibility highlighting the role of autophagy in controlling infection, inflammation and cancer [13,15,83,84]. Furthermore, gene mutation or deletion studies have indicated that the autophagy pathway affects the onset and exacerbation of IBD via several mechanisms including clearance of invading bacteria, secretion of granules from Paneth cells, inflammasome activity, pro-inflammatory cytokine production and endoplasmic reticulum (ER) stress. However, the role of autophagy in the pathogenesis of IBD is still debated. Although many researchers focused on the involvement of ATGs in IBD pathogenesis, little is known about the autophagic role of ATGs and the mechanisms that confer intestinal inflammation. Table 1 summarizes the ATGs and transcription factors described in this review and their functional relationships in intestinal pathogenesis.
4.1. ATG16L1
Numerous studies have reported that genetic variation in ATG16L1 is associated with IBD risk in ethnically diverse populations [13,14,92,93,94,95,96]. Notably, the rs2241880 single nucleotide polymorphism (SNP; T300A) of ATG16L1 was repeatedly found in several Caucasian cohorts, suggesting a strong association of this variant with the incidence of CD, although it was not frequently found in other populations, particularly in Asian patients [13,84,96].
ATG16L1, a homolog of ATG16, is essential in the formation of autophagosomes, along with the ATG12-ATG5 conjugate [97,98]. Importantly, Cadwell et al. showed that mice with low expression of ATG16L1 (ATG16L1HM mice) exhibited abnormal Paneth cell granule secretion and that mice with ATG16L1 deficiency in Paneth cells had a defect in the granule exocytosis pathway [76]. Similarly, patients carrying the ATG16L1 risk allele (T300A) had pathological features such as disorganized granules or diffuse Paneth cell cytoplasmic lysozyme staining [76]. Using IEC-specific ATG16L1-deficient mice and ex vivo IEC organoids, a recent study showed that ATG16L1 in IECs played an essential role in controlling pathology, intestinal inflammation and TNF-induced apoptosis [86]. Additionally, previous studies showed that the ER stress sensor inositol-requiring enzyme (IRE)-1α accumulated in Paneth cells of ATG16L1ΔIEC mice and CD patients (T300A), suggesting that defective autophagy leads to pathological activation of IRE1α to drive intestinal inflammation [85]. Moreover, loss of IKKα function markedly impaired the secretion of cytoprotective IL-18 and upregulated ER stress responses through decreased ATG16L1 stabilization [99]. These data emphasize the role of ER stress in defective ATG16L1-mediated colonic inflammation [85,99]. Indeed, IEC-specific deletion of ATG16L1 or ATG7 led to hyper-activated ER stress, which may amplify the severity of intestinal inflammation in autophagy-defective conditions [100].
Although IECs, particularly Paneth cells, are important in defective ATG16L1-associated intestinal inflammation, the function of ATG16L1 in myeloid cells has also been demonstrated [87]. Saitoh et al. showed that ATG16L1-deficient macrophages exhibited Toll/IL-1 receptor domain-containing adaptor inducing interferon (IFN)-β (TRIF)-dependent activation of the inflammasome, resulting in the production of high amounts of the inflammatory cytokines IL-1β and IL-18 [87]. Deficiency of ATG16L1 in hematopoietic cells resulted in an increased susceptibility to dextran sulfate sodium (DSS)-induced colitis, suggesting an essential role for ATG16L1 in the control of intestinal inflammation [87]. Another study using mice with myeloid ATG16L1 deficiency showed exacerbated colitis with upregulated proinflammatory responses as well as increased colitogenic bacteria, indicating that ATG16L1 deficiency results in alterations in macrophage function that affect the severity of CD [88].
Peripheral blood mononuclear cells (PBMCs) isolated from CD patients with the ATG16L1 T300A risk variant have been shown to exhibit increased production of the proinflammatory cytokines IL-1β and IL-6, particularly in response to NOD2 ligands [101]. Moreover, the loss of ATG16L1 increased TRIF and its signaling, resulting in increased production of type I IFN and IL-1β [102]. Interestingly, the genetic variant ATG16L1 T300A was found to be associated with adalimumab treatment, suggesting that this SNP affects the response to treatment with immunomodulatory drugs [103]. Importantly, the CD risk allele T300A variant (T316A in mice) is associated with accelerated degradation of ATG16L1 due to caspase-3 activation. Upon apoptotic stimuli or metabolic stress, human and murine macrophages harboring T300A or T316A variants of ATG16L1, respectively, exhibited accelerated degradation of ATG16L1, leading to decreased autophagy, defective clearance of the pathogen and enhanced inflammation [104]. These data strongly suggest that the functional defect in ATG16L1 is involved in the dysregulation of intestinal homeostasis and CD pathogenesis (Figure 3).
4.2. IRGM
Human immunity-related guanosine triphosphatase (GTPase) family M (IRGM) encodes the only functional immunity-related GTPase (IRG) among IRG family members [105]. Involvement of genetic polymorphisms of IRGM in CD and tuberculosis has been previously demonstrated, particularly in GWASs [16,106,107]. A meta-analysis showed that the IRGM variants rs13361189 and rs4958847 are associated with both UC and CD in human IBD [12]. However, another study in a Korean population showed that selected SNPs of IRGM were associated with CD but not UC susceptibility [17].
Human IRGM (syn: LRG47, IFI1), which is encoded by the immunity-related GTPase protein family, M gene (IRGM; 5q33.1), is thought to be distant from a class of IRGs in mice. There are more than 20 IRG genes (IRGM1–3, IRGN1–8, IRGB1–10 and IRGD) in mice, whereas there is only one IRGM gene present in humans and chimpanzees, making the study of the role of IRGs in vivo difficult. Earlier studies showed that the murine GTPase IRGM1 (LRG-47) was important for autophagy activation to eliminate intracellular Mycobacterium tuberculosis [108,109,110]. A further study showed that a human IRG protein, the human ortholog IRGM1 (IRGM), contributed to the control of M. tuberculosis through autophagy activation [111]. Tiwari et al. reported an essential function of Irgm1 as an innate effector in targeting the mycobacterial phagosome through lipid-mediated binding to enhance phagosome maturation and the antimicrobial response [112]. Furthermore, IRGM has been shown to regulate autophagy by translocating to the mitochondria and influencing mitochondrial fission, which is required for autophagic defense against intracellular mycobacteria [113]. Studies using IRG-deficient mice showed that IRGM can be induced by IFN-γ and plays a role in the clearance of intracellular bacteria including Toxoplasma gondii, Listeria monocytogenes and Salmonella spp. as well as mycobacteria (Figure 4) [110,114,115,116,117,118]. Human IRGM and murine IRGM1 contribute to cell-autonomous defense though autophagy activation via the recruitment of both autophagic and SNARE adaptor proteins during infection (Figure 4) [18,110,111,117,119,120,121,122]. However, IRGM favors viral replication through autophagy activation. For example, IRGM is translocated to the Golgi apparatus, where it regulates Golgi membrane fragmentation and is involved in virus-triggered autophagy activation during hepatitis C infection [123].
Murine and human studies have demonstrated the protective role of IRGM in the maintenance of intestinal homeostasis. Irgm1-deficient mice have been shown to exhibit functional defects in intestinal Paneth cells and hyperinflammation in the colon and ileum following chemical exposure [124]. In addition, the IRGM protein contributed to the limitation of CD-associated intracellular AIEC in epithelial cells through autophagy activation and phagosomal maturation [121]. In the intestinal mucosa, greater quantities of pathogenic AIEC, which invade IECs and induce TNF-α, are found in CD patients than in healthy controls [72,125]. These data collectively suggest the importance of IRGM in CD pathogenesis via limitation of pathogenic bacteria through autophagy activation. A recent cohort study revealed the relationship among autophagy-related IRGM variants, visceral adipose tissue and nonalcoholic fatty liver disease, which shows an increased morbidity with CD [126]. However, there is still a debate regarding the relevance of autophagy in CD in terms of IRGM, as autophagy activation has been observed in Paneth cells in CD patients, independently of IRGM variants associated with CD susceptibility [127]. Moreover, RNA analysis showed that most autophagy gene sets were downregulated by appendectomy, which contributed to protection against UC [128]. Suppression of autophagy may offer cross-reactive immunity between host antigens and microbes through decreased antigen processing, thereby ameliorating symptoms of colitis [128].
4.3. LRRK2/MUC19
LRRK2/MUC19 is a complex protein that contains a RAS of complex proteins (ROC) GTPase domain, a C-terminal ROC domain and a Ser/Thr kinase domain and is involved in NOD2-mediated signaling, of which autophagy is a downstream process [129]. Because LRRK2 is a well-known gene involved in the pathogenesis of Parkinson’s disease (PD), most earlier studies were performed in neuronal cells [130,131]. Later, meta-GWASs identified the links between LRRK2 and CD and leprosy, suggesting a role for LRRK2 in immune regulation during infection and inflammation [80,132,133]. LRRK2 is highly expressed in myeloid cells and B cells which is induced by IFN-γ and is involved in the production of inflammatory cytokines and antimicrobial responses in macrophages [80,132,133]. In addition, LRRK2 is required for commensal bacteria-driven cargo sorting through recruitment to lysozyme-containing dense core vesicles in Paneth cells, thereby participating in the coordination of the lysozyme-sorting process in the intestine to promote symbiosis [134].
A previous genome-wide linkage analysis suggested that a locus on chromosome 12 (historically known as PARK8) is linked to familial parkinsonism in the Japanese population [135]. Further studies have demonstrated the involvement of LRRK2 in autosomal-dominant parkinsonism in multiple families [130,131]. In addition, two meta-GWASs reported LRRK2 as a CD susceptibility gene [79,80]. A genome-wide conjunctional analysis revealed several novel loci, which are potentially involved in the association between PD and autoimmune diseases [136]. For example, known PD loci adjacent to LRRK2 (rs17467164) were proposed as overlapping susceptibility loci for UC and CD [136]. Accumulating data in conjunction with the development of in silico analyses may identify novel genetic variants that affect the risk of several diseases occurring in combinatorial patterns. In a Japanese CD cohort, a defective Paneth cell phenotype was correlated with clinical characteristics and autophagy-associated LRRK2 (LRRK2M2397T) was associated with Paneth cell defects [22]. The majority of LRRK2 SNPs, which are associated with IBD, are found in non-coding intronic regions [24]. There is speculation that there might be a relationship between the high frequency of non-coding region SNPs in LRRK2 and the stability/expression levels of LRRK2 [24].
Several studies have investigated the mechanistic aspects of pathogenic LRRK2 in PD models (Figure 5). Pathogenic LRRK2 is involved in protein translation through regulation of microRNA function (let-7 and miR-184*), which results in altered production of E2F1/DP and is critical for cell cycle and survival [137]. The autosomal dominant mutant protein LRRK2 phosphorylates and activates transcription of the forkhead box transcription factor FoxO1, which is crucial in the upregulation of cell death molecules and is associated with LRRK2-mediated cell death [138]. Importantly, LRRK2 deficiency led to impairment of the autophagy-lysosomal pathway, altered expression of LC3-II and p62 and increased α-synuclein aggregates in the kidney, in an age-dependent manner [139]. Recent studies have shown that mitochondrial RHOT1-dependent mitophagy is delayed with the PD mutant LRRK2G2019S, suggesting a critical function of LRRK2 in the regulation of mitophagy [140]. Although LRRK2 is known to be involved in autophagic flux, the exact roles and mechanisms by which LRRK2 controls intestinal homeostasis are not completely understood in terms of autophagy regulation [129,139].
The molecular mechanisms underlying how LRRK2 affects the pathogenesis of CD have not been widely examined. An earlier study showed that LRRK2 deficiency led to increased susceptibility to DSS-induced colitis in mouse models by negatively regulating activation of the transcription factor NFAT [23]. In a recent study, both lymphoblastoid cells from control patients bearing a high-risk allele of LRRK2 and dendritic cells from CD patients exhibited elevated LRRK2 expression, which resulted in severe colitis with increased Dectin-1-mediated NF-κB activation and proinflammatory cytokine responses [25]. As an IFN-γ target gene, LRRK2 induction and function were investigated in immune cells [133,141]. LRRK2 was previously detected in inflamed intestinal tissues, particularly in macrophages of the lamina propria and was shown to play a role in host defense through regulation of reactive oxygen species generation [133]. These studies collectively suggest that the fine-tuning of LRRK2 is required for the prevention and treatment of colitis and related infections (Figure 5).
4.4. ATG7
ATG7 is an E1-like activating enzyme that facilitates autophagosome formation through two ubiquitin-like conjugation systems, LC3 lipidation and Atg12 conjugation. Availability of ATG conditional deletion mice have improved our understanding of the contribution of different ATGs in specific cells/tissues and provided insight into the role of individual ATGs in intestinal homeostasis during colitis. In intestinal cells, the function of Atg7 has been studied using intestinal epithelium-specific (tamoxifen-inducible) Atg7 knockout (ATG7IEC-KO) mice. An earlier study showed that ATG7IEC-KO mice had a similar pathology in the ileum and Paneth cell abnormality with defective granule exocytosis as those observed in Atg16L1HM and Atg5flox/floxvillin-Cre mice [142]. Consistent with these observations, ATG7IEC-KO mice exhibited decreased granule size and decreased lysozyme levels in Paneth cells and increased production of TNF-α and IL-1β mRNA in response to lipopolysaccharide in the epithelium of the small intestine when compared to those of control small intestinal tissue [143,144]. A further study showed that the exacerbated experimental colitis in ATG7IEC-KO mice was associated with abnormal microflora composition and dysregulated expression of antimicrobial or antiparasitic peptides (angiogenin-4, Relmβ, intelectin-1 and intelectin-2), as well as suppressed secretion of colonic mucins [145].
ATG7 conditional knockout mice also exhibited increased susceptibility to and reduced clearance of Citrobacter rodentium infection in the intestinal epithelium during C. rodentium infectious colitis [89]. A recent study emphasized the role of autophagy in controlling intestinal homeostasis using mice with conditionally deleted ATG7 in CD11c+ antigen-presenting cells (ATG7ΔAPC), which enhanced immunopathology and inflammatory Th17 responses, as well as abnormal mitochondrial function and oxidative stress [90]. Another group showed that mice with myeloid cell-specific deletion of ATG7 exhibited increased susceptibility to experimental colitis accompanied with increased colonic inflammation [91]. Furthermore, ATG7 deletion in intestinal epithelium-specific XBP1-deficient mice synergistically aggravated the intestinal pathology, resulting in the development of extensive submucosal or transmural inflammation and recapitulated the features of human CD, suggesting that autophagy contributes to a compensatory process in the intestinal epithelium during sustained ER stress [100]. These data strongly indicate that ATG7 is crucial for controlling intestinal inflammatory responses and defense against the virulence of enteric pathogens to maintain intestinal homeostasis (Figure 2). However, there is little evidence that ATG7 is clinically relevant in IBD.
5. Selective Autophagic Receptors and IBD
The involvement of the autophagic receptors p62 and TFEB in IBD pathogenesis has been reported [21,146]. Several studies have also identified SNPs of NDP52 and OPTN in individuals with IBD [20,147,148]. Here, we discuss the current evidence regarding the role of these cargo receptors in terms of IBD.
5.1. p62
The ubiquitin-binding protein sequestosome 1 (SQSTM1/p62) is a well-known autophagy adaptor and was initially identified as a p56lck binding protein [149]. Mutations of p62 are known to be associated with Paget’s disease of bone [150]. An earlier study showed that a direct interaction between p62 and LC3/GABARAP family members led to autophagy-mediated destruction of p62-positive, polyubiquitin-containing bodies [151]. In addition, Komatsu et al. showed the function of p62 in selective autophagy activation via the binding of ubiquitinylated protein aggregates for delivery to LC3 autophagosomes [152]. In canonical autophagy, accumulation of p62 in the cytoplasm is generally regarded as a sign of reduced autophagic activity and the impaired autophagy, because increased autophagic flux degrades p62 (Figure 2) [153]. Overall, p62 is an essential scaffold protein that can bind a variety of partner proteins, participating in diverse biological signaling that affects innate immunity, apoptosis, inflammatory responses and tumorigenesis [154].
Although there are a few reports on the involvement of p62 in the pathogenesis of colitis, defective autophagy with decreased turnover of p62 levels has been observed in intestinal inflammation [155,156]. In a study performed in an epithelial cell line, the intracellular survival of AIEC LF82 bacteria, which promote the gastrointestinal inflammatory response, was higher in cells silenced for p62 than in cells transduced with empty vector (ShCTR) [157]. In animal and human studies, defective autophagic flux with elevated p62 was observed in IBD tissues and models [156]. Furthermore, immunohistochemical expression of p62 was higher in epithelial cells of damaged mucosa than in those of non-damaged mucosa [146]. Understanding how p62 regulates intestinal homeostasis will enable the development of more effective therapeutic strategies against IBD.
5.2. Optineurin (OPTN)
OPTN is a selective autophagy adaptor protein that plays an important role in mitophagy and xenophagy. OPTN is involved in various biological responses including vesicular trafficking, anti-bacterial and antiviral responses and autophagy and interacts with numerous cellular proteins including myosin VI, Rab8 and Tank-binding kinase 1 [158,159,160]. The clinical relevance of OPTN has been indicated by the genetic variants/mutations of OPTN linked to glaucoma, Paget’s disease of bone and amyotrophic lateral sclerosis [161,162,163]. OPTN plays an essential role in mitophagy and is involved in several neurodegenerative diseases, including PD [160,164]. OPTN is also required for the clearance of pathogens, such as Salmonella and Listeria, to promote activation of xenophagy [43,165].
A few studies have identified OPTN deficiency in CD patients [19,20]. The function of OPTN in the regulation of intestinal homeostasis was suggested in IRE1α-driven colitis as an IRE1α-interacting protein [85]. OPTN deficiency led to an accumulation of IRE1α, which enhanced colitis pathology during ER stress (Figure 2) [85]. Indeed, a subgroup of CD patients with low OPTN expression has been identified [20]. The same study showed that OPTN deficiency led to decreased production of TNFα and IL-6 and increased susceptibility to Citrobacter colitis and E. coli peritonitis (Figure 2) [19]. Thus, these data suggest the importance of OPTN in macrophage inflammation and bactericidal function to promote the antimicrobial host defense and may explain the link between OPTN deficiency and increased CD pathogenesis [19]. These insights indicate that selective autophagy and cargo receptors may play important roles in the regulation of colonic homeostasis (Figure 2). A better understanding of the players and mechanisms underlying selective autophagy in intestinal inflammation will aid in the discovery of new therapeutic targets for IBD.
6. Transcription Factor TFEB
TFEB, a member of the microphthalmia-associated transcription factor (MITF)/transcriptional factor E (TFE) family, has been identified as a key regulator of autophagy maturation and lysosome biogenesis [166,167,168,169]. TFEB activation is required for clearance of pathogenic molecular aggregates in neurodegenerative diseases, such as α-synuclein and aberrant tau protein, to promote therapeutic effects in PD and Alzheimer’s disease, respectively [170,171,172].
Our understanding of the effects of TFEB in IBD is currently in its infancy. A recent study showed that mice with a conditional deletion of TFEB in the intestinal epithelium (TFEBΔIEC) had a defect in Paneth cell granules, lower expression levels of lipoprotein ApoA1 and exaggerated colitis upon DSS injury [21]. Further studies evaluating the function of TFEB and its clinical relevance in IBD pathogenesis are needed, given the essential role of TFEB in regulating the autophagy lysosome pathway [168]. Elucidation of the involvement of TFEB and other transcription factors in colitis will improve our understanding of the mechanism of autophagic regulation in the complicated pathogenesis of IBD.
7. Conclusions
Studies over the last decade have suggested that genetic variants of several ATGs are highly associated with IBD susceptibility. Although the distinct genetic variations or manipulations provide strong support for the causative role of ATGs in the pathogenesis of IBD, they also raise two important questions: whether defective autophagy is a major trigger for pathogenic inflammation in IBD and what signaling pathways control canonical and noncanonical autophagy in intestinal epithelial cells/tissues. We focused our review on ATGs, selective autophagy receptors and transcription factors involved in maintaining intestinal homeostasis in human and mouse studies.
Both ATG16L1 and IRGM, two important ATGs in IBD, have principally been investigated in human and murine intestinal epithelial cells, particularly in Paneth cells, the major secretory cells of the small intestine. Recent studies have suggested that defective ATG16L1-mediated inflammation is due to aberrant ER stress, as upregulated IRE1α was observed in Paneth cells from ATG16L1ΔIEC mice and CD patients (T300A). The interactions between ATGs and other biological systems such as the ER stress response may have a significant impact on the pathogenesis of IBD as well as other inflammatory diseases. Furthermore, genetic association studies have suggested that LRRK2/MUC19 and ATG7 deficiency aggravate intestinal inflammation in a mouse model of colitis.
Selective autophagy receptors, p62 and OPTN and the transcription factor TFEB have been suggested to play key roles in controlling intestinal inflammation and homeostasis. Future studies to further define the mechanisms by which cargo receptors contribute to specific types of autophagy will enhance our understanding of intestinal homeostasis in terms of autophagy regulation. This knowledge will ultimately aid in the development of novel therapeutic strategies and drug targets for combating intractable chronic inflammatory diseases such as IBD.
Funding
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIP) (No. 2017R1A5A2015385), by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. NRF-2015M3C9A2054326) and by research fund of Chungnam National University.
Acknowledgments
We thank Y.S.K. and P. Silwal for critical discussion and reading of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Chen, X.W.; Liu, C. Intestinal Autophagy and Its Pharmacological Control in Inflammatory Bowel Disease. Front. Immunol. 2016, 7, 695. [Google Scholar] [CrossRef]
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Muzes, G.; Tulassay, Z.; Sipos, F. Interplay of autophagy and innate immunity in Crohn’s disease: A key immunobiologic feature. World J. Gastroenterol. 2013, 19, 4447–4454. [Google Scholar] [CrossRef]
- Fritz, T.; Niederreiter, L.; Adolph, T.; Blumberg, R.S.; Kaser, A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut 2011, 60, 1580–1588. [Google Scholar] [CrossRef]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Pareja, M.E.; Colombo, M.I. Autophagic clearance of bacterial pathogens: Molecular recognition of intracellular microorganisms. Front. Cell. Infect. Microbiol. 2013, 3, 54. [Google Scholar] [CrossRef]
- Bah, A.; Vergne, I. Macrophage Autophagy and Bacterial Infections. Front. Immunol. 2017, 8, 1483. [Google Scholar] [CrossRef]
- Fesus, L.; Demeny, M.A.; Petrovski, G. Autophagy shapes inflammation. Antioxid. Redox Signal. 2011, 14, 2233–2243. [Google Scholar] [CrossRef]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef]
- Palomino-Morales, R.J.; Oliver, J.; Gomez-Garcia, M.; Lopez-Nevot, M.A.; Rodrigo, L.; Nieto, A.; Alizadeh, B.Z.; Martin, J. Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: A meta-analysis approach. Genes Immun. 2009, 10, 356–364. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Lakatos, P.L.; Szamosi, T.; Szilvasi, A.; Molnar, E.; Lakatos, L.; Kovacs, A.; Molnar, T.; Altorjay, I.; Papp, M.; Tulassay, Z.; et al. ATG16L1 and IL23 receptor (IL23R) genes are associated with disease susceptibility in Hungarian CD patients. Dig. Liver Dis. 2008, 40, 867–873. [Google Scholar] [CrossRef]
- Salem, M.; Ammitzboell, M.; Nys, K.; Seidelin, J.B.; Nielsen, O.H. ATG16L1: A multifunctional susceptibility factor in Crohn disease. Autophagy 2015, 11, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Moon, C.M.; Shin, D.J.; Kim, S.W.; Son, N.H.; Park, A.; Park, B.; Jung, E.S.; Kim, E.S.; Hong, S.P.; Kim, T.I.; et al. Associations between genetic variants in the IRGM gene and inflammatory bowel diseases in the Korean population. Inflamm. Bowel Dis. 2013, 19, 106–114. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Huett, A.; Kuballa, P.; Chilewski, S.D.; Landry, A.; Goyette, P.; Zody, M.C.; Hall, J.L.; Brant, S.R.; Cho, J.H.; et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet. 2008, 40, 1107–1112. [Google Scholar] [CrossRef]
- Chew, T.S.; O’Shea, N.R.; Sewell, G.W.; Oehlers, S.H.; Mulvey, C.M.; Crosier, P.S.; Godovac-Zimmermann, J.; Bloom, S.L.; Smith, A.M.; Segal, A.W. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Dis. Models Mech. 2015, 8, 817–829. [Google Scholar] [CrossRef]
- Smith, A.M.; Sewell, G.W.; Levine, A.P.; Chew, T.S.; Dunne, J.; O’Shea, N.R.; Smith, P.J.; Harrison, P.J.; Macdonald, C.M.; Bloom, S.L.; et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology 2015, 144, 45–55. [Google Scholar] [CrossRef]
- Murano, T.; Najibi, M.; Paulus, G.L.C.; Adiliaghdam, F.; Valencia-Guerrero, A.; Selig, M.; Wang, X.; Jeffrey, K.; Xavier, R.J.; Lassen, K.G.; et al. Transcription factor TFEB cell-autonomously modulates susceptibility to intestinal epithelial cell injury in vivo. Sci. Rep. 2017, 7, 13938. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.C.; Naito, T.; Liu, Z. LRRK2 but not ATG16L1 is associated with Paneth cell defect in Japanese Crohn’s disease patients. JCI Insight 2017, 2, e91917. [Google Scholar] [CrossRef]
- Liu, Z.; Lee, J.; Krummey, S.; Lu, W.; Cai, H.; Lenardo, M.J. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 2011, 12, 1063–1070. [Google Scholar] [CrossRef]
- Liu, Z.; Lenardo, M.J. The role of LRRK2 in inflammatory bowel disease. Cell Res. 2012, 22, 1092–1094. [Google Scholar] [CrossRef]
- Takagawa, T.; Kitani, A. An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Kuballa, P.; Nolte, W.M.; Castoreno, A.B.; Xavier, R.J. Autophagy and the immune system. Annu. Rev. Immunol. 2012, 30, 611–646. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Klionsky, D.J. Regulation of autophagy: Modulation of the size and number of autophagosomes. FEBS Lett. 2014, 588, 2457–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertollano, R.; Ferguson, S.M. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumura, K.; Choi, A.M.; Ryter, S.W. Emerging role of selective autophagy in human diseases. Front. Pharmacol. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Simonsen, A. The elimination of accumulated and aggregated proteins: A role for aggrephagy in neurodegeneration. Neurobiol. Dis. 2011, 43, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef] [Green Version]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yao, Z.; Klionsky, D.J. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Mercer, C.A.; Kaliappan, A.; Dennis, P.B. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009, 5, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-monitoring and autophagy-deficient mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif—Crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, D.K. Inflammatory bowel disease (1). N. Engl. J. Med. 1991, 325, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.C.; Shaker, A.; Levin, M.S. Chronic intestinal inflammation: Inflammatory bowel disease and colitis-associated colon cancer. Front. Immunol. 2012, 3, 107. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.K.; Kwak, J.E.; Shin, E.C. IL-17A-Producing Foxp3(+) Regulatory T Cells and Human Diseases. Immune Netw. 2017, 17, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Mowat, C.; Cole, A.; Windsor, A.; Ahmad, T.; Arnott, I.; Driscoll, R.; Mitton, S.; Orchard, T.; Rutter, M.; Younge, L.; et al. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011, 60, 571–607. [Google Scholar] [CrossRef]
- Kim, D.H.; Cheon, J.H. Pathogenesis of Inflammatory Bowel Disease and Recent Advances in Biologic Therapies. Immune Netw. 2017, 17, 25–40. [Google Scholar] [CrossRef]
- Elphick, D.A.; Mahida, Y.R. Paneth cells: Their role in innate immunity and inflammatory disease. Gut 2005, 54, 1802–1809. [Google Scholar] [CrossRef]
- Noah, T.K.; Donahue, B.; Shroyer, N.F. Intestinal development and differentiation. Exp. Cell Res. 2011, 317, 2702–2710. [Google Scholar] [CrossRef] [Green Version]
- Bry, L.; Falk, P.; Huttner, K.; Ouellette, A.; Midtvedt, T.; Gordon, J.I. Paneth cell differentiation in the developing intestine of normal and transgenic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 10335–10339. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Leblond, C.P. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am. J. Anat. 1974, 141, 537–561. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, A.J. Paneth cells and innate mucosal immunity. Curr. Opin. Gastroenterol. 2010, 26, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cederlund, A.; Gudmundsson, G.H.; Agerberth, B. Antimicrobial peptides important in innate immunity. FEBS J. 2011, 278, 3942–3951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.M.; Stamp, G.W.; Elia, G.; Poulsom, R.; Wright, N.A. Proliferative populations in intestinal metaplasia: Evidence of deregulation in Paneth and goblet cells, but not endocrine cells. J. Pathol. 2000, 190, 107–113. [Google Scholar] [CrossRef]
- Gunther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 2011, 477, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Hodin, C.M.; de Haan, J.J.; Derikx, J.P.; Rouschop, K.M.; Verheyen, F.K.; van Dam, R.M.; Dejong, C.H.; Buurman, W.A.; Lenaerts, K. Level of activation of the unfolded protein response correlates with Paneth cell apoptosis in human small intestine exposed to ischemia/reperfusion. Gastroenterology 2011, 140, 529–539.e523. [Google Scholar] [CrossRef]
- Rubio, C.A. Lysozyme-rich mucus metaplasia in duodenal crypts supersedes Paneth cells in celiac disease. Virchows Arch. 2011, 459, 339–346. [Google Scholar] [CrossRef]
- Paulus, G.L.; Xavier, R.J. Autophagy and checkpoints for intracellular pathogen defense. Curr. Opin. Gastroenterol. 2015, 31, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Gardet, A.; Xavier, R.J. Common alleles that influence autophagy and the risk for inflammatory bowel disease. Curr. Opin. Immunol. 2012, 24, 522–529. [Google Scholar] [CrossRef]
- Salzman, N.H.; Bevins, C.L. Dysbiosis--a consequence of Paneth cell dysfunction. Semin. Immunol. 2013, 25, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S.; Gordon, J.I.; Glimcher, L.H. Homeostasis and inflammation in the intestine. Cell 2010, 140, 859–870. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nunez, G.; Flavell, R.A. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005, 307, 731–734. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef]
- de Lange, K.M.; Barrett, J.C. Understanding inflammatory bowel disease via immunogenetics. J. Autoimmun. 2015, 64, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef] [Green Version]
- Brest, P.; Corcelle, E.A.; Cesaro, A.; Chargui, A.; Belaid, A.; Klionsky, D.J.; Vouret-Craviari, V.; Hebuterne, X.; Hofman, P.; Mograbi, B. Autophagy and Crohn’s disease: At the crossroads of infection, inflammation, immunity, and cancer. Curr. Mol. Med. 2010, 10, 486–502. [Google Scholar] [CrossRef] [PubMed]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W. Defective ATG16L1-mediated removal of IRE1alpha drives Crohn’s disease-like ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Kabat, A.M.; Maloy, K.J. Intestinal Epithelial Cell Autophagy Is Required to Protect against TNF-Induced Apoptosis during Chronic Colitis in Mice. Cell Host Microbe 2018, 23, 191–202.e194. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, L.; McGovern, D.P.; Hamill, A.M. Myeloid ATG16L1 Facilitates Host-Bacteria Interactions in Maintaining Intestinal Homeostasis. J. Immunol. 2017, 198, 2133–2146. [Google Scholar] [CrossRef]
- Inoue, J.; Nishiumi, S.; Fujishima, Y.; Masuda, A.; Shiomi, H.; Yamamoto, K.; Nishida, M.; Azuma, T.; Yoshida, M. Autophagy in the intestinal epithelium regulates Citrobacter rodentium infection. Arch. Biochem. Biophys. 2012, 521, 95–101. [Google Scholar] [CrossRef]
- Ravindran, R.; Loebbermann, J.; Nakaya, H.I.; Khan, N.; Ma, H.; Gama, L.; Machiah, D.K.; Lawson, B.; Hakimpour, P.; Wang, Y.C.; et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature 2016, 531, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Kim, J.; Quan, W.; Lee, J.C.; Kim, M.S.; Kim, S.H.; Bae, J.W.; Hur, K.Y.; Lee, M.S. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy 2016, 12, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.R.; Cooney, R.; Pathan, S.; Anderson, C.A.; Barrett, J.C.; Beckly, J.; Geremia, A.; Hancock, L.; Guo, C.; Ahmad, T.; et al. Confirmation of the role of ATG16L1 as a Crohn’s disease susceptibility gene. Inflamm. Bowel Dis. 2007, 13, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Prescott, N.J.; Fisher, S.A.; Franke, A.; Hampe, J.; Onnie, C.M.; Soars, D.; Bagnall, R.; Mirza, M.M.; Sanderson, J.; Forbes, A.; et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology 2007, 132, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Onouchi, Y.; Takazoe, M.; Kubo, M.; Nakamura, Y.; Hata, A. Association analysis of genetic variants in IL23R, ATG16L1 and 5p13.1 loci with Crohn’s disease in Japanese patients. J. Hum. Genet. 2007, 52, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.L.; Gearry, R.B.; Hollis-Moffatt, J.E.; Miller, A.L.; Reid, J.; Abkevich, V.; Timms, K.M.; Gutin, A.; Lanchbury, J.S.; Merriman, T.R.; et al. IL23R R381Q and ATG16L1 T300A are strongly associated with Crohn’s disease in a study of New Zealand Caucasians with inflammatory bowel disease. Am. J. Gastroenterol. 2007, 102, 2754–2761. [Google Scholar] [CrossRef]
- Girardelli, M.; Basaldella, F.; Paolera, S.D.; Vuch, J.; Tommasini, A.; Martelossi, S.; Crovella, S.; Bianco, A.M. Genetic profile of patients with early onset inflammatory bowel disease. Gene 2018, 645, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef] [Green Version]
- Diamanti, M.A.; Gupta, J. IKKalpha controls ATG16L1 degradation to prevent ER stress during inflammation. J. Exp. Med. 2017, 214, 423–437. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Plantinga, T.S.; Crisan, T.O.; Oosting, M.; van de Veerdonk, F.L.; de Jong, D.J.; Philpott, D.J.; van der Meer, J.W.; Girardin, S.E.; Joosten, L.A.; Netea, M.G. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 2011, 60, 1229–1235. [Google Scholar] [CrossRef]
- Samie, M.; Lim, J.; Verschueren, E.; Baughman, J.M.; Peng, I.; Wong, A.; Kwon, Y.; Senbabaoglu, Y.; Hackney, J.A. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat. Immunol. 2018, 19, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Nuij, V.; Peppelenbosch, M.P.; van der Woude, C.J.; Fuhler, G.M. Genetic polymorphism in ATG16L1 gene is associated with adalimumab use in inflammatory bowel disease. J. Transl. Med. 2017, 15, 248. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; DeVoss, J.; Diehl, L.; Graham, R.R.; et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy in infection. Curr. Opin. Cell Biol. 2010, 22, 252–262. [Google Scholar] [CrossRef]
- Chauhan, S.; Mandell, M.A.; Deretic, V. Mechanism of action of the tuberculosis and Crohn disease risk factor IRGM in autophagy. Autophagy 2016, 12, 429–431. [Google Scholar] [CrossRef]
- Fisher, S.A.; Tremelling, M.; Anderson, C.A.; Gwilliam, R.; Bumpstead, S.; Prescott, N.J.; Nimmo, E.R.; Massey, D.; Berzuini, C.; Johnson, C.; et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat. Genet. 2008, 40, 710–712. [Google Scholar] [CrossRef]
- Hunn, J.P.; Feng, C.G.; Sher, A.; Howard, J.C. The immunity-related GTPases in mammals: A fast-evolving cell-autonomous resistance system against intracellular pathogens. Mamm. Genome 2011, 22, 43–54. [Google Scholar] [CrossRef]
- Bekpen, C.; Marques-Bonet, T.; Alkan, C.; Antonacci, F.; Leogrande, M.B.; Ventura, M.; Kidd, J.M.; Siswara, P.; Howard, J.C.; Eichler, E.E. Death and resurrection of the human IRGM gene. PLoS Genet. 2009, 5, e1000403. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Choi, H.P.; Matsuzawa, T.; Pypaert, M.; MacMicking, J.D. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P(2) and PtdIns(3,4,5)P(3) promotes immunity to mycobacteria. Nat. Immunol. 2009, 10, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Ornatowski, W.; Vergne, I.; Naylor, J.; Delgado, M.; Roberts, E.; Ponpuak, M.; Master, S.; Pilli, M.; White, E.; et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 2010, 12, 1154–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, G.A.; Feng, C.G.; Sher, A. p47 GTPases: Regulators of immunity to intracellular pathogens. Nat. Rev. Immunol. 2004, 4, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Henry, S.C.; Daniell, X.; Indaram, M.; Whitesides, J.F.; Sempowski, G.D.; Howell, D.; Oliver, T.; Taylor, G.A. Impaired macrophage function underscores susceptibility to Salmonella in mice lacking Irgm1 (LRG-47). J. Immunol. 2007, 179, 6963–6972. [Google Scholar] [CrossRef] [PubMed]
- Collazo, C.M.; Yap, G.S.; Sempowski, G.D.; Lusby, K.C.; Tessarollo, L.; Vande Woude, G.F.; Sher, A.; Taylor, G.A. Inactivation of LRG-47 and IRG-47 reveals a family of interferon gamma-inducible genes with essential, pathogen-specific roles in resistance to infection. J. Exp. Med. 2001, 194, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Shenoy, A.R.; Kumar, P.; Bradfield, C.J.; MacMicking, J.D. IFN-inducible GTPases in host cell defense. Cell Host Microbe 2012, 12, 432–444. [Google Scholar] [CrossRef]
- Kim, B.H.; Shenoy, A.R.; Kumar, P.; Das, R.; Tiwari, S.; MacMicking, J.D. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science 2011, 332, 717–721. [Google Scholar] [CrossRef]
- Traver, M.K.; Henry, S.C.; Cantillana, V.; Oliver, T.; Hunn, J.P.; Howard, J.C.; Beer, S.; Pfeffer, K.; Coers, J.; Taylor, G.A. Immunity-related GTPase M (IRGM) proteins influence the localization of guanylate-binding protein 2 (GBP2) by modulating macroautophagy. J. Biol. Chem. 2011, 286, 30471–30480. [Google Scholar] [CrossRef]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hebuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef]
- Lapaquette, P.; Glasser, A.L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef]
- Kumar, S.; Jain, A. Mechanism of Stx17 recruitment to autophagosomes via IRGM and mammalian Atg8 proteins. J. Cell Biol. 2018, 217, 997–1013. [Google Scholar] [CrossRef]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 114, E3462–E3471. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef] [Green Version]
- Rolhion, N.; Darfeuille-Michaud, A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 1277–1283. [Google Scholar] [CrossRef]
- Simon, T.G.; Van Der Sloot, K.W.J.; Chin, S.B.; Joshi, A.D.; Lochhead, P.; Ananthakrishnan, A.N.; Xavier, R.; Chung, R.T.; Khalili, H. IRGM Gene Variants Modify the Relationship Between Visceral Adipose Tissue and NAFLD in Patients With Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 2247–2257. [Google Scholar] [CrossRef]
- Thachil, E.; Hugot, J.P.; Arbeille, B.; Paris, R.; Grodet, A.; Peuchmaur, M.; Codogno, P.; Barreau, F.; Ogier-Denis, E.; Berrebi, D.; et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn’s disease. Gastroenterology 2012, 142, 1097–1099.e1094. [Google Scholar] [CrossRef]
- Cheluvappa, R.; Luo, A.S.; Grimm, M.C. Autophagy suppression by appendicitis and appendectomy protects against colitis. Inflamm. Bowel Dis. 2014, 20, 847–855. [Google Scholar] [CrossRef]
- Toledo Pinto, T.G.; Batista-Silva, L.R.; Medeiros, R.C.A.; Lara, F.A.; Moraes, M.O. Type I Interferons, Autophagy and Host Metabolism in Leprosy. Front. Immunol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef]
- Zhang, F.R.; Huang, W.; Chen, S.M.; Sun, L.D.; Liu, H.; Li, Y.; Cui, Y.; Yan, X.X.; Yang, H.T.; Yang, R.D.; et al. Genomewide association study of leprosy. N. Engl. J. Med. 2009, 361, 2609–2618. [Google Scholar] [CrossRef]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef]
- Zhang, Q.; Pan, Y.; Yan, R.; Zeng, B.; Wang, H.; Zhang, X.; Li, W.; Wei, H.; Liu, Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat. Immunol. 2015, 16, 918–926. [Google Scholar] [CrossRef]
- Funayama, M.; Hasegawa, K.; Kowa, H.; Saito, M.; Tsuji, S.; Obata, F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol. 2002, 51, 296–301. [Google Scholar] [CrossRef]
- Witoelar, A.; Jansen, I.E.; Wang, Y.; Desikan, R.S.; Gibbs, J.R.; Blauwendraat, C.; Thompson, W.K.; Hernandez, D.G.; Djurovic, S.; Schork, A.J.; et al. Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 2017, 74, 780–792. [Google Scholar] [CrossRef]
- Gehrke, S.; Imai, Y.; Sokol, N.; Lu, B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 2010, 466, 637–641. [Google Scholar] [CrossRef]
- Chuang, C.L.; Lu, Y.N.; Wang, H.C.; Chang, H.Y. Genetic dissection reveals that Akt is the critical kinase downstream of LRRK2 to phosphorylate and inhibit FOXO1, and promotes neuron survival. Hum. Mol. Genet. 2014, 23, 5649–5658. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J., 3rd; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef]
- Wang, X. Destructive cellular paths underlying familial and sporadic Parkinson disease converge on mitophagy. Autophagy 2017, 13, 1998–1999. [Google Scholar] [CrossRef]
- Kuss, M.; Adamopoulou, E.; Kahle, P.J. Interferon-gamma induces leucine-rich repeat kinase LRRK2 via extracellular signal-regulated kinase ERK5 in macrophages. J. Neurochem. 2014, 129, 980–987. [Google Scholar] [CrossRef]
- Cadwell, K.; Patel, K.K.; Komatsu, M.; Virgin, H.W.t.; Stappenbeck, T.S. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 2009, 5, 250–252. [Google Scholar] [CrossRef]
- Wittkopf, N.; Gunther, C.; Martini, E.; Waldner, M.; Amann, K.U.; Neurath, M.F.; Becker, C. Lack of intestinal epithelial atg7 affects paneth cell granule formation but does not compromise immune homeostasis in the gut. Clin. Dev. Immunol. 2012, 2012, 278059. [Google Scholar] [CrossRef]
- Fujishima, Y.; Nishiumi, S.; Masuda, A.; Inoue, J.; Nguyen, N.M.; Irino, Y.; Komatsu, M.; Tanaka, K.; Kutsumi, H.; Azuma, T.; et al. Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-kappaB activation. Arch. Biochem. Biophys. 2011, 506, 223–235. [Google Scholar] [CrossRef]
- Tsuboi, K.; Nishitani, M.; Takakura, A.; Imai, Y.; Komatsu, M.; Kawashima, H. Autophagy Protects against Colitis by the Maintenance of Normal Gut Microflora and Secretion of Mucus. J. Biol. Chem. 2015, 290, 20511–20526. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Masia, D.; Cosin-Roger, J.; Calatayud, S.; Hernandez, C.; Alos, R.; Hinojosa, J.; Apostolova, N.; Alvarez, A.; Barrachina, M.D. Hypoxic macrophages impair autophagy in epithelial cells through Wnt1: Relevance in IBD. Mucosal Immunol. 2014, 7, 929–938. [Google Scholar] [CrossRef]
- Till, A.; Lipinski, S.; Ellinghaus, D.; Mayr, G.; Subramani, S.; Rosenstiel, P.; Franke, A. Autophagy receptor CALCOCO2/NDP52 takes center stage in Crohn disease. Autophagy 2013, 9, 1256–1257. [Google Scholar] [CrossRef] [Green Version]
- Ellinghaus, D.; Zhang, H.; Zeissig, S.; Lipinski, S.; Till, A.; Jiang, T.; Stade, B.; Bromberg, Y.; Ellinghaus, E.; Keller, A.; et al. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology 2013, 145, 339–347. [Google Scholar] [CrossRef]
- Joung, I.; Strominger, J.L.; Shin, J. Molecular cloning of a phosphotyrosine-independent ligand of the p56lck SH2 domain. Proc. Natl. Acad. Sci. USA 1996, 93, 5991–5995. [Google Scholar] [CrossRef]
- Laurin, N.; Brown, J.P.; Morissette, J.; Raymond, V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 2002, 70, 1582–1588. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. BioMed Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef]
- Cosin-Roger, J.; Simmen, S.; Melhem, H.; Atrott, K.; Frey-Wagner, I.; Hausmann, M.; de Valliere, C.; Spalinger, M.R.; Spielmann, P.; Wenger, R.H.; et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat. Commun. 2017, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Paiva, N.M.; Pascoal, L.B.; Negreiros, L.M.V.; Portovedo, M.; Coope, A.; Ayrizono, M.L.S.; Coy, C.S.R.; Milanski, M.; Leal, R.F. Ileal pouch of ulcerative colitis and familial adenomatous polyposis patients exhibit modulation of autophagy markers. Sci. Rep. 2018, 8, 2619. [Google Scholar] [CrossRef]
- Mimouna, S.; Bazin, M.; Mograbi, B.; Darfeuille-Michaud, A.; Brest, P.; Hofman, P.; Vouret-Craviari, V. HIF1A regulates xenophagic degradation of adherent and invasive Escherichia coli (AIEC). Autophagy 2014, 10, 2333–2345. [Google Scholar] [CrossRef]
- Sundaramoorthy, V.; Walker, A.K.; Tan, V.; Fifita, J.A.; McCann, E.P.; Williams, K.L.; Blair, I.P.; Guillemin, G.J.; Farg, M.A.; Atkin, J.D. Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 3452. [Google Scholar] [CrossRef]
- Ying, H.; Yue, B.Y. Optineurin: The autophagy connection. Exp. Eye Res. 2016, 144, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Albagha, O.M.; Visconti, M.R.; Alonso, N.; Langston, A.L.; Cundy, T.; Dargie, R.; Dunlop, M.G.; Fraser, W.D.; Hooper, M.J.; Isaia, G.; et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat. Genet. 2010, 42, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.Y.; Beyens, G.; Boonen, S.; Papapoulos, S.; Geusens, P.; Karperien, M.; Vanhoenacker, F.; Verbruggen, L.; Fransen, E.; Van Offel, J.; et al. The majority of the genetic risk for Paget’s disease of bone is explained by genetic variants close to the CSF1, OPTN, TM7SF4, and TNFRSF11A genes. Hum. Genet. 2010, 128, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, A.J.; Pallanck, L.J. PINK1/Parkin mitophagy and neurodegeneration-what do we really know in vivo? Curr. Opin. Genet. Dev. 2017, 44, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Puri, M.; La Pietra, L.; Mraheil, M.A.; Lucas, R.; Chakraborty, T.; Pillich, H. Listeriolysin O Regulates the Expression of Optineurin, an Autophagy Adaptor That Inhibits the Growth of Listeria monocytogenes. Toxins (Basel) 2017, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Polito, V.A.; Li, H.; Martini-Stoica, H.; Wang, B.; Yang, L.; Xu, Y.; Swartzlander, D.B.; Palmieri, M.; di Ronza, A.; Lee, V.M.; et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 2014, 6, 1142–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Overview of autophagy, selective autophagy and ATGs (A) Molecular machinery of autophagy process. After mTOR inhibition or AMPK activation, the autophagy process begins with the biogenesis of the phagophore/isolation membrane. The ATG16L1-ATG5-ATG12 and LC3-II-PE conjugates participate in autophagosome formation process. The mature autophagosomes are fused with a late endosome and lysosome to initiate degradation of cargos. Finally, cells recycle the released products in cytosol. (B) Selective autophagy clears various targets such as subcellular structure, bacteria, protein and lipid aggregates.
Figure 1.
Overview of autophagy, selective autophagy and ATGs (A) Molecular machinery of autophagy process. After mTOR inhibition or AMPK activation, the autophagy process begins with the biogenesis of the phagophore/isolation membrane. The ATG16L1-ATG5-ATG12 and LC3-II-PE conjugates participate in autophagosome formation process. The mature autophagosomes are fused with a late endosome and lysosome to initiate degradation of cargos. Finally, cells recycle the released products in cytosol. (B) Selective autophagy clears various targets such as subcellular structure, bacteria, protein and lipid aggregates.

Figure 2.
The process of autophagy flux and involved genes. The autophagy flux is depicted. A normal autophagic flux includes the autophagosome formation and maturation step and the autolysosome formation step. The possible conditions associated with involved genes are depicted: (1) OPTN deficiency leads to an accumulation of IRE1α and increased susceptibility of Citrobacter and E. coli. (2) ATG7 deletion is associated with increased inflammation. (3) TFEB deletion results in increased inflammation and lysosomal defect.
Figure 2.
The process of autophagy flux and involved genes. The autophagy flux is depicted. A normal autophagic flux includes the autophagosome formation and maturation step and the autolysosome formation step. The possible conditions associated with involved genes are depicted: (1) OPTN deficiency leads to an accumulation of IRE1α and increased susceptibility of Citrobacter and E. coli. (2) ATG7 deletion is associated with increased inflammation. (3) TFEB deletion results in increased inflammation and lysosomal defect.

Figure 3.
Summary diagram showing the role of ATG16L1 in the Crohn’s disease. The left panel demonstrates the normal host defense mechanism against intracellular pathogens. Healthy cells exhibit normal granule secretion, autophagic activity, ER stress response and permeability. The right panel shows the ATG16L1 T300A variant cells defective in granule secretion, autophagy process, IRE1α degradation and tight junction barrier function.
Figure 3.
Summary diagram showing the role of ATG16L1 in the Crohn’s disease. The left panel demonstrates the normal host defense mechanism against intracellular pathogens. Healthy cells exhibit normal granule secretion, autophagic activity, ER stress response and permeability. The right panel shows the ATG16L1 T300A variant cells defective in granule secretion, autophagy process, IRE1α degradation and tight junction barrier function.

Figure 4.
Autophagy targets adherent-invasive Escherichia coli (AIEC), Mycobacteria, Salmonella and Listeria by different mechanisms. Stimulation with IFN-γ induce IRGM to clear intracellular bacteria. Furthermore, IRGM can be induced by IFN-γ contribute to cell-autonomous defense though autophagy activation via the recruitment of both autophagic and SNARE adaptor proteins during infection.
Figure 4.
Autophagy targets adherent-invasive Escherichia coli (AIEC), Mycobacteria, Salmonella and Listeria by different mechanisms. Stimulation with IFN-γ induce IRGM to clear intracellular bacteria. Furthermore, IRGM can be induced by IFN-γ contribute to cell-autonomous defense though autophagy activation via the recruitment of both autophagic and SNARE adaptor proteins during infection.

Figure 5.
The pathogenic roles of LRRK in non-intestine and intestine diseases. In Parkinson’s disease, LRRK2 is involved in cell cycle/survival, ROS generation, autophagolysosomal pathway, mitophagy and inflammation. LRRK2 plays a key role in intestine homeostasis through regulating NFAT, NF-κB, ROS generation, inflammation and autophagy.
Figure 5.
The pathogenic roles of LRRK in non-intestine and intestine diseases. In Parkinson’s disease, LRRK2 is involved in cell cycle/survival, ROS generation, autophagolysosomal pathway, mitophagy and inflammation. LRRK2 plays a key role in intestine homeostasis through regulating NFAT, NF-κB, ROS generation, inflammation and autophagy.


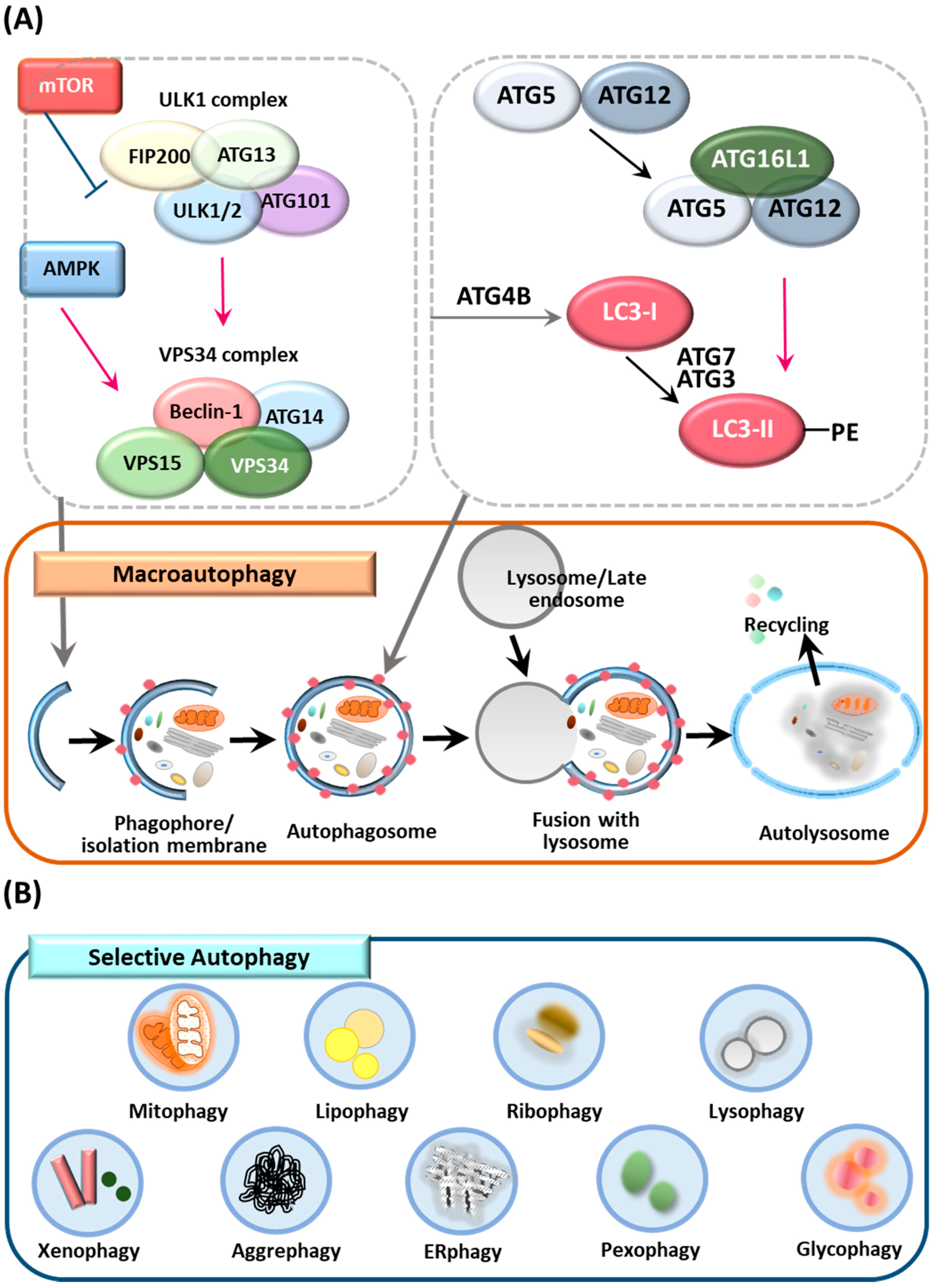{kind=link}
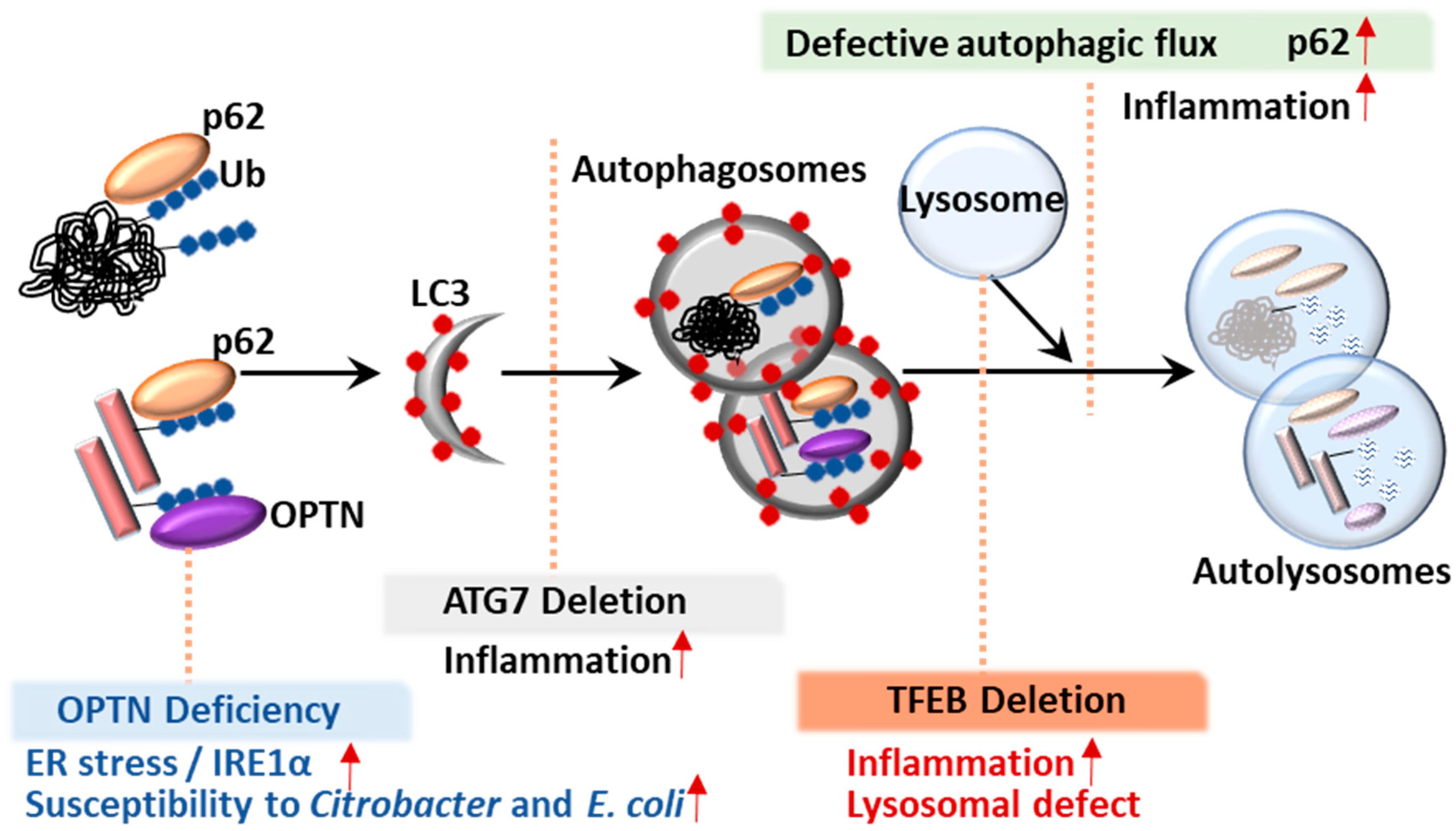{kind=link}
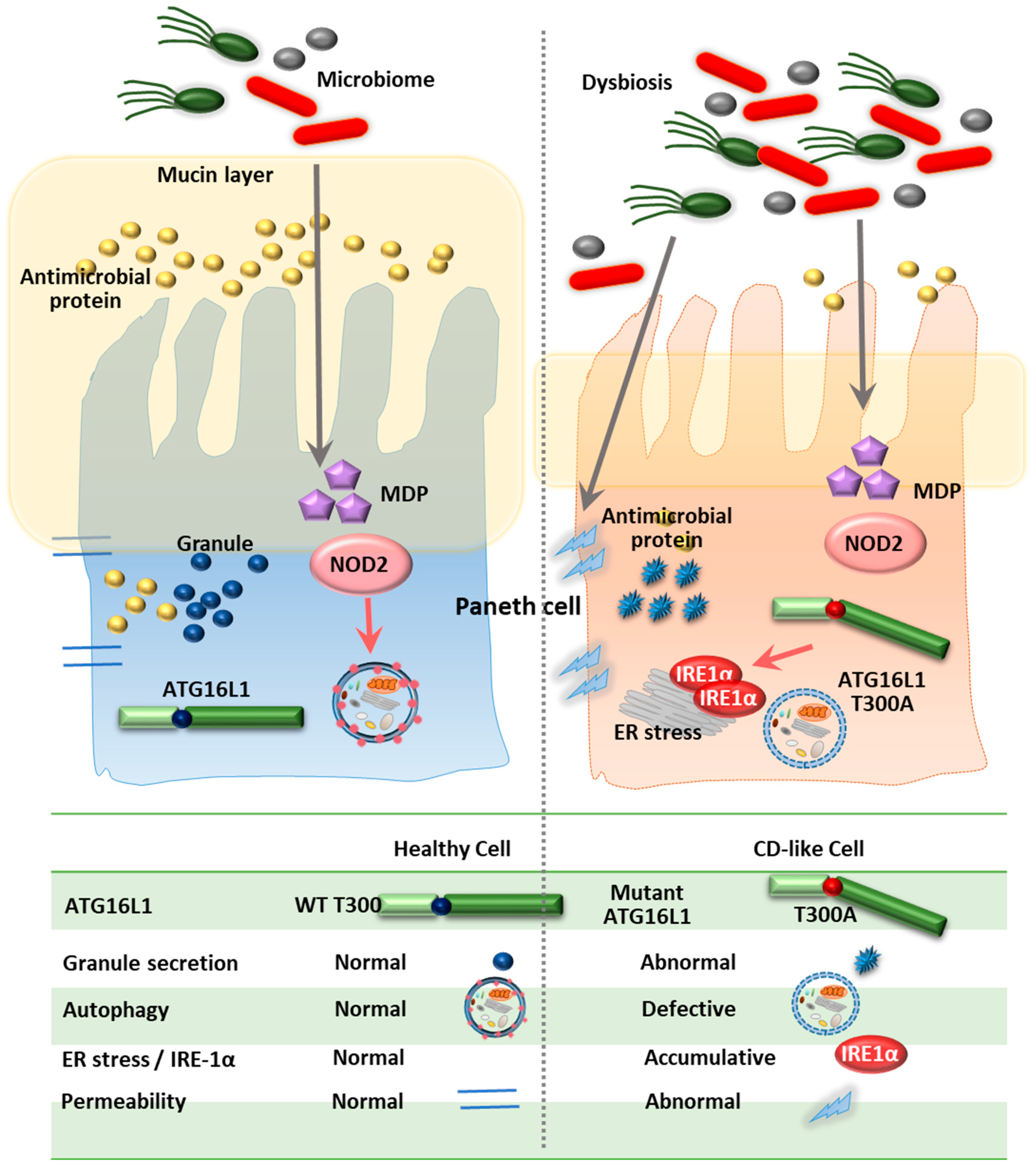{kind=link}
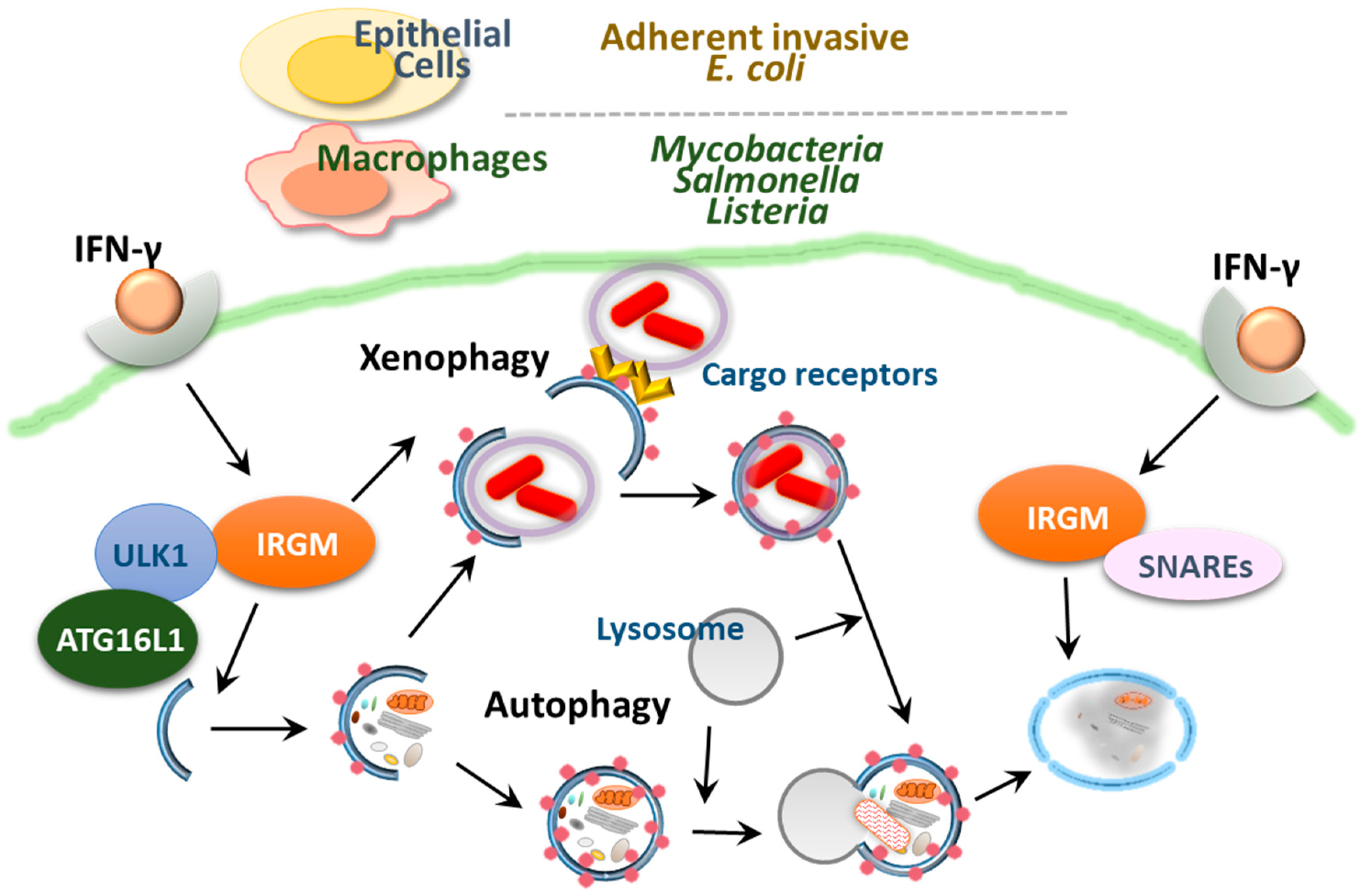{kind=link}
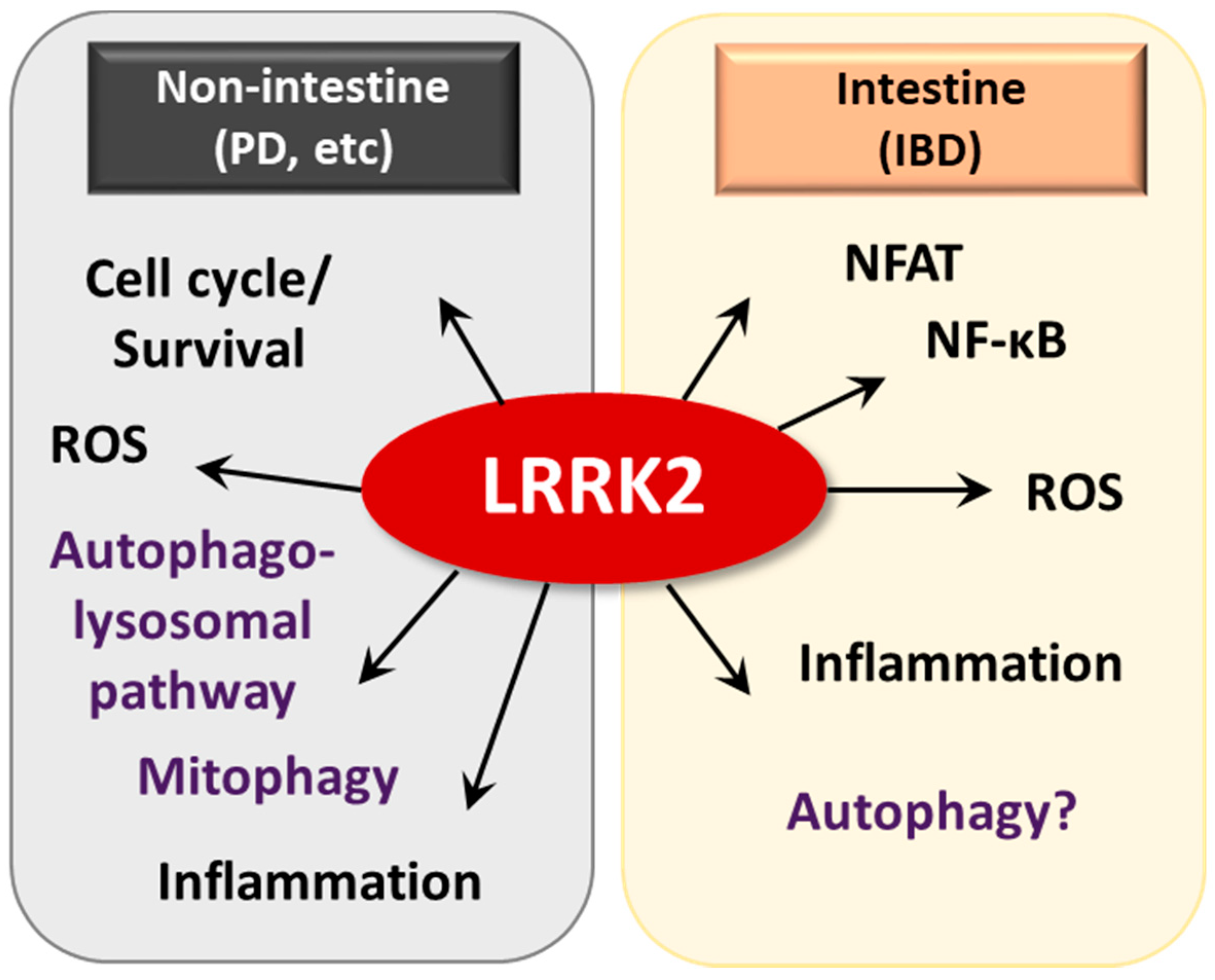{kind=link}
Table 1.
Genetic models related to autophagy in intestinal pathogenesis.
| Knocked-Out Gene | Cell Type | Mechanism | Outcome | Reference |
|---|---|---|---|---|
| ATG16L1 | Intestinal epithelial cells | Abnormality of Paneth cell granule secretion and defect in the granule exocytosis pathway | [76] | |
| Intestinal epithelial cells | ER stress sensor inositol-requiring enzyme (IRE)-1α accumulated in Paneth cells | Increased intestinal inflammation | [85] | |
| Intestinal epithelial cells | Elevated pro-inflammatory cytokine secretion and increased IEC apoptosis after Helicobacter hepaticus infection | Exacerbated murine model of chronic colitis | [86] | |
| Myeloid cells | Production high amounts of the inflammatory cytokines IL-1β and IL-18 via Toll/IL-1 receptor domain-containing adaptor inducing interferon (IFN)-β (TRIF)-dependent activation of the inflammasome | Increased susceptibility to dextran sulfate sodium (DSS)-induced colitis | [87] | |
| Myeloid cells | Increased reactive oxygen species production, impaired mitophagy, reduced microbial killing, impaired processing of MHC class II Ags and altered intracellular trafficking to the lysosomal compartments | Exacerbated murine model of acute and chronic colitis | [88] | |
| Myeloid cells | No effect on disease severity in murine model of chronic colitis | [86] | ||
| IRGM | Intestinal epithelial cells | Marked alterations of Paneth cell location and granule morphology | Hyperinflammation in the colon and ileum following chemical exposure | [85] |
| LRRK2/ MUC19 | Myeloid cells | Activation of the transcription factor NFAT | Increased susceptibility to DSS-induced colitis in mouse models | [23] |
| ATG7 | Intestinal epithelial cells | Higher expression levels of pro-inflammatory cytokine mRNA in the large intestine after infection | Increased susceptibility to Citrobacter rodentium infectious colitis in mouse models | [89] |
| Intestinal antigen presenting cells | Enhanced immunopathology and inflammatory Th17 responses, as well as abnormal mitochondrial function and oxidative stress | Increased susceptibility to DSS-induced colitis in mouse models | [90] | |
| Myeloid cells | Increased colonic cytokine expression, T helper 1 skewing and systemic bacterial invasion | Increased susceptibility to DSS-induced colitis in mouse models | [91] | |
| OPTN | Myeloid cells | Decreased antimicrobial host defense (decreased production of TNFα and IL-6) after infection | Increased susceptibility to Citrobacter colitis and E. coli peritonitis | [19] |
| TFEB | Intestinal epithelial cells | Defect in Paneth cell granules, lower expression levels of lipoprotein ApoA1 | Exaggerated colitis upon DSS injury | [21] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, S.; Eun, H.S.; Jo, E.-K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 2019, 8, 77. https://doi.org/10.3390/cells8010077
AMA Style
Kim S, Eun HS, Jo E-K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells. 2019; 8(1):77. https://doi.org/10.3390/cells8010077
Chicago/Turabian StyleKim, Sup, Hyuk Soo Eun, and Eun-Kyeong Jo. 2019. "Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease" Cells 8, no. 1: 77. https://doi.org/10.3390/cells8010077
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.