Yeast to Study Human Purine Metabolism Diseases



1
Université de Bordeaux IBGC UMR 5095 1, rue Camille Saint-Saëns, F-33077 Bordeaux, France
2
Centre National de la Recherche Scientifique IBGC UMR 5095 1, rue Camille Saint-Saëns, F-33077 Bordeaux, France
*
Author to whom correspondence should be addressed.
Cells 2019, 8(1), 67; https://doi.org/10.3390/cells8010067
Submission received: 8 January 2019
/
Revised: 15 January 2019
/
Accepted: 15 January 2019
/
Published: 17 January 2019
(This article belongs to the Special Issue Simple Organisms for Complex Problems: Modeling Human Disease in Yeast and Dictyostelium)
Abstract
:Purine nucleotides are involved in a multitude of cellular processes, and the dysfunction of purine metabolism has drastic physiological and pathological consequences. Accordingly, several genetic disorders associated with defective purine metabolism have been reported. The etiology of these diseases is poorly understood and simple model organisms, such as yeast, have proved valuable to provide a more comprehensive view of the metabolic consequences caused by the identified mutations. In this review, we present results obtained with the yeast Saccharomyces cerevisiae to exemplify how a eukaryotic unicellular organism can offer highly relevant information for identifying the molecular basis of complex human diseases. Overall, purine metabolism illustrates a remarkable conservation of genes, functions and phenotypes between humans and yeast.
1. Introduction
Purine nucleotides, adenosine 5’-triphosphate (ATP), guanosine 5’-triphosphate (GTP) and their derivatives are involved in a myriad of cellular processes: energy storage, synthesis of nucleic acids and coenzymes (Nicotinamide adenine dinucleotide (NAD)/Nicotinamide adenine dinucleotide phosphate (NADP)/coenzyme A/flavine adenine dinucleotide (FAD)), translation, signaling, etc. These molecules are thus absolutely required for all known forms of life, and their synthesis results essentially from conserved pathways. In yeast, mutations abolishing ATP or GTP synthesis are lethal, although lethality may require more than one mutation due to genetic and pathway redundancy [1]. Even a partial block of purine metabolism can have drastic physiological consequences, and several diseases associated with purine metabolism dysfunctions have been reported in human [2,3,4,5]. Some purine metabolic disorders have been described for a long time, such as hyperuricemia (gout), which is caused by an excess of uric acid (the final purine degradation product) leading to a painful deposit of urate crystals in joints. Among the studies of genetic alterations leading to hyperuricemia, hypoxanthine phosphoribosyl transferase (HGPRT)-deficiency, involved in the Lesch–Nyhan syndrome, was one of the very first genetic-disease enzymes identified in humans [6]. In addition to hyperuricemia, purine metabolism-associated diseases share a large spectrum of immunological, hematological and neuro-muscular disorders [7], and are all characterized by an abnormal level of purine nucleotides in cells and of nucleosides and/or nucleobases in bodily fluids [8]. In most cases, the dysfunctional gene in purine metabolism is known. However, this identification of the causative enzyme does not necessarily give clear indications on the etiology of the disease and hence, a more comprehensive view of the metabolic consequences of the dysfunction is often needed. To this end, model organisms that are amenable to genetics can be valuable for identifying critical functions that are affected as a consequence of the primary metabolic dysfunction. Several animal or microbial models can be used for this purpose and are often highly complementary. In this review, we present results obtained with the budding yeast, Saccharomyces cerevisiae, to illustrate how a unicellular eukaryotic organism can offer highly relevant information to help understand the mechanisms leading to complex human diseases.
Yeast can be both a source of information and a tool used to study human metabolic diseases. In both cases, the relevance of the information drawn from yeast depends on the functional conservation between yeast and humans, a conservation which is often much higher than one could have initially thought based on their long divergent evolution (over one billion years). In a systematic replacement of yeast genes by their human orthologues, Marcotte and coworkers showed that nearly half of the yeast genes could be successfully “humanized” [9], thus confirming the wide potential of yeast as a model to study diseases associated to human gene dysfunctions. As a unicellular eukaryotic organism yeast is a very polyvalent tool, since it is highly amenable to molecular genetics but also to analytical biochemistry. Studies on yeast have provided a multitude of information on metabolic pathways and how they are connected with one another as well as to other cellular functions. In addition, as an organism facing nutrient changeability and stress [1], it also delivered key information on how metabolic homeostasis is achieved in complex systems. This integrated view offered by yeast research is of great value to help address complex metabolic diseases in human. Indeed, most diseases are the result of multiple effects and take place in a highly integrated genetic and physiological context. Knowledge collected from yeast can be used to generate testable hypotheses in human cells or model animals.
In this review, we will focus on purine metabolic diseases and illustrate how yeast can be used to help promote our understanding of the mechanisms involved. As stated above, yeast can be used as a model provided that sufficient functional conservation is found. However, to be highly informative as a model, yeast should demonstrate both conservation of enzymes and pathways, as well as a conserved functional organization including interactions between the metabolic pathways. This last aspect cannot be generalized and requires thorough specific investigations. Several examples developed below illustrate the remarkable level of conservation in the “logic” of metabolism and in the consequences of its dysfunctions. This suggests that the general mechanisms responsible for nucleotide homeostasis are very ancient and that their conservation has been under high selective pressure.
2. Purine Metabolism in Human and Yeast: Similarities and Differences
Purine Metabolic Pathways: Functions Are Generally Conserved but Protein Sequences Can Diverge
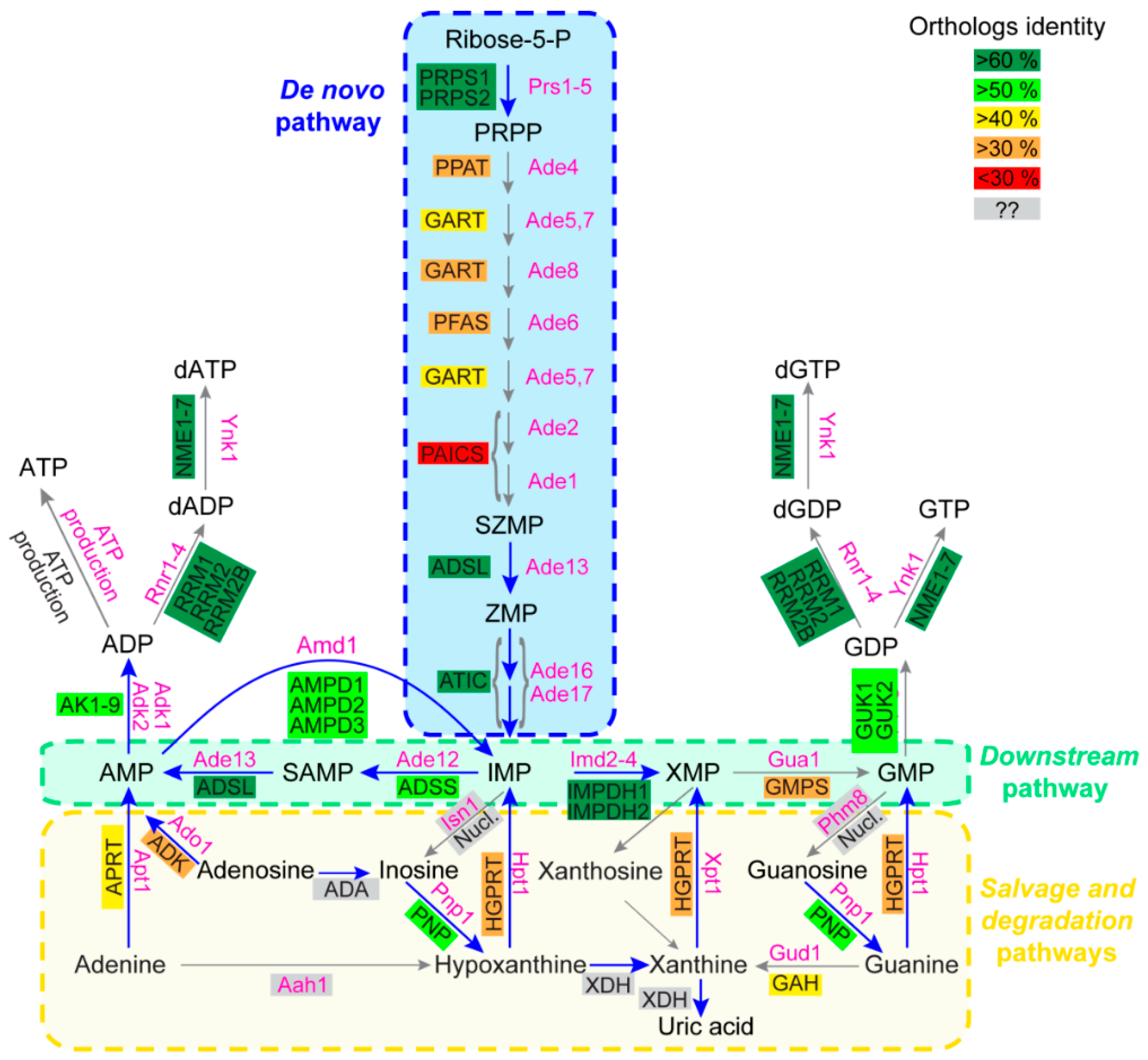
Most enzymatic steps of the purine de novo and recycling pathways are catalyzed by proteins that are largely conserved between prokaryotes and eukaryotes [1]. Accordingly, a high degree of conservation is found between yeast and human purine pathways. The ten enzymatic steps of the de novo pathway are fully conserved, although the percentage of identity among orthologous enzymes is variable, as illustrated in Figure 1. Remarkably, the four most conserved proteins (>60% identity between orthologs) in the de novo and downstream pathways are mutated in reported diseases (Figure 1, blue arrows), i.e., Phosphoribosyl pyrophosphate (PRPP) synthetase (PRPS1); adenylosuccinate lyase (ADSL), aminoimidazole carboxamide riboside monophosphate transformylase inosine 5’-monophosphate cyclohydrolase (ATIC) and IMP dehydrogenase (IMPDH) (Table 1). Of note, both the ADSL and ATIC steps metabolize intermediates acting as regulators of various functions in both yeast and humans (see below, Section 3.2) [10,11,12,13,14]. A lower identity is detected for proteins catalyzing the seven other steps of the de novo pathway, and several of these enzymes are encoded as gene fusions in the human genome (see GART and PAICS, Figure 1). For the purine salvage pathway, conservation varies between the nucleotide monophosphate interconversion enzymes and the nucleoside/nucleobase salvagers. Indeed, the interconversion of monophosphate nucleotides is ensured by highly conserved proteins, while enzymes metabolizing nucleosides and nucleobases show little to no conservation (Figure 1). Among those enzymes, the first group corresponds to proteins that are highly divergent in sequence but for which catalytic activity is nevertheless conserved. These proteins correspond to the phosphoribosyl transferases (APRT and HGPRT, Figure 1), which catalyze nucleotide monophosphate synthesis from nucleobases, and to the nucleosidases (Phm8 [15] and Isn1 [16] in yeast and the NT5 nucleotidase family in human [17]) that catabolize nucleotide monophosphate to nucleosides. The second group contains proteins with no orthologs between yeast and humans. For example, in humans, adenosine is deaminated into inosine by ADA [18] with no yeast ortholog and, by contrast, the yeast adenine deaminase (Aah1) catalyzing adenine to hypoxanthine conversion [19] has no ortholog in human cells (Figure 1). Of note, this difference in the evolution of nucleobase and nucleoside metabolism in yeast versus humans is strengthened by the fact that nucleobases are the only purine precursors taken up by yeast [1] via the concentrative carrier Fcy2 [20], while nucleobases and nucleosides are both transported into human cells by a set of equilibrative (hENT) and concentrative (hCNT) membrane transporters [21]. Another major difference observed between yeast and humans relates to purine degradation. In humans, nucleobases are transformed to uric acid by xanthine oxidoreductases [8], while in yeast, removal of purine excess is performed by nucleobase excretion (essentially hypoxanthine) [13,22,23]. Indeed, no xanthine oxidase activity has been reported in yeast. By contrast, the other purine degradation steps which are common to both yeast and humans correspond to purine nucleoside phosphorylase (inosine to hypoxanthine transformation by PNP/Pnp1) and guanine deaminase (guanine to xanthine metabolization by GAH/Gud1), which are both well conserved between yeast and humans (Figure 1).
In summary, this comparison highlights that the purine-associated human diseases correspond to alterations in the most conserved steps of the purine pathways (Figure 1, blue arrows). When altered, these steps are also detrimental in yeast and belong to the following pathways: (1) The monophosphate interconversion pathway which is common to the de novo and salvage pathways (downstream pathway), which allows interconversion between purines and the balanced synthesis of the final products ATP and GTP (Figure 1), (2) the phosphoribosyl pyrophosphate (PRPP) synthesis which is required for both the de novo pathway (for PPAT/Ade4) and the salvage pathway (for the phosphoribosyl transferases APRT and HGPRT) and (3) purine degradation (via PNP/Pnp1). In most of these purine-associated diseases, however, it is not clear whether the detrimental effects are linked to a toxic accumulation of the substrate(s) of these enzymes, to a lack of their products, or to a combination of both effects. The high degree of conservation observed between the two organisms allows us to use yeast genetics to address these issues and raise hypotheses testable in human cells or small animal models.
3. Yeast: A Model to Tackle Complex Purine-Associated Diseases
Metabolic genetic diseases most often affect metabolic enzymes which in the mutant form is either inactive (loss of function), hyperactive or inadequately regulated. These dysfunctions can result in different metabolic consequences which are not at all exclusive:
- Important metabolites may not be synthesized, or synthesized insufficiently
- Metabolites, which are accumulated due to an increased synthesis or the lack of metabolizing enzymes, can be toxic because they interfere with other processes (this could be particularly true for metabolites which have physiological regulatory properties)
- A metabolic unbalance can occur, which is often due to a mix of the first two hypotheses
In many instances, mutations in yeast and human orthologues not only result in the same biochemical dysfunction (typically loss of activity) but also may lead to highly similar secondary consequences. Below, using various examples, we will illustrate the issues raised by complex metabolic diseases and how yeast can help to address these.
3.1. AMP-Deaminase (AMPD2) Deficiency Associated with Pontocerebellar Hypoplasia Results in Defective ATP/GTP Balance
AMP-deaminase is an important purine interconversion enzyme, which allows synthesis of IMP from AMP (Figure 1). In yeast there is a unique isoform encoded by the AMD1 gene, while in humans there are three isoforms expressed in different tissues. Deficiency of the muscular form, AMPD1, is associated with hyper-fatigability and is a relatively frequent mutation, although it is often asymptomatic, which suggests complex interplays with other factors [8]. The lack of the yeast enzyme was associated with an increased ATP and low GTP under conditions where ATP was synthesized from adenine. In the meantime, the nucleotide balance was unaffected when amd1-deleted yeast cells were grown under conditions where ATP was synthesized from IMP [33]. This conditional phenotype lead to the conclusion that the low GTP was a result of ATP accumulation, which occurred through an ATP feedback inhibition of Ade4, the first enzyme of the de novo pathway [13]. This allosteric inhibition resulted in a lowered IMP synthesis and consequently a low intracellular GMP and GTP [33]. More recently, the identification in humans of AMPD2 deficiency as the cause of pontocerebellar hypoplasia in a cohort of patients revealed that the exact same phenomenon operated in human cells [34]. These authors showed that, just as in yeast, the GTP shortage in AMPD2 deficient human cells was dependent on replenishment of the adenylic nucleotide pool by a purine precursor (adenosine for human cells, adenine for yeast) [33,34]. In both cases, ATP accumulation resulted in a low GTP, through regulatory means. Thus, in the AMP deaminase-deficient cells, ATP was toxic by affecting the ATP/GTP balance in both yeast and human cells. Accordingly, restoring intracellular GTP through guanine or AICAR feeding in yeast and human cells, respectively, was sufficient enough to abolish ATP toxicity [33,34]. This illustrated how a single mutation could cause a complex phenotype through shortage of the reaction product (IMP), accumulation of the substrate (AMP) and the resulting imbalance of downstream metabolic products (ATP and GTP).
In their study, Akizu and coworkers also took advantage of yeast to functionally validate AMPD2 as an AMP deaminase by complementation of the amd1 growth defect specifically on adenine. They also expressed Human AMPD2 mutant forms in yeast cells and showed that these alleles resulted in poorly functional enzymes [34]. Finally, these authors identified translation initiation as a defect resulting from GTP shortage, in both humans and yeast, and proposed that it could contribute to the etiology of this disease [34]. Importantly, once again yeast was used to document this phenomenon in depth [34]. This study thus nicely illustrates not only how information derived from yeast genetics can be used to characterize a human disease, but also how yeast can be used as a tool to functionally validate the various alleles of a human gene or to study molecular mechanisms that could cause the disease. The authors proposed that the dependence on adenosine for expression of the phenotype could explain the neural-specificity of the defect, since a significant amount of adenosine was present in the brain [35]. Hence, “brainless” yeast proved to be very useful for understanding this neurodegenerative disorder. More generally, this work exemplifies the remarkable conservation of genes, functions, phenotypes and mechanisms between human and yeast.
3.2. Deficiencies in the Purine De Novo Pathway: Toxic Accumulation of Metabolic Intermediates?
Strikingly, while a succession of 13 enzymatic steps are required for AMP de novo synthesis, so far mutations in only three of the corresponding genes have been identified as associated with diseases. These three genes encode phosphoribosyl pyrophosphate synthase (PRPS) [29,30,31,32], adenylosuccinate lyase (ADSL) [36] and AICAR-transformylase IMP cyclohydrolase (ATIC) [25]. The reasons why no disease has ever been associated with mutations in the other steps of the de novo pathway are not known, but it is remarkable that SZMP (Succinyl Amino Imidazole Carboxamide Ribonucleotide monophosphate) and ZMP (Amino Imidazole Carboxamide ribonucleotide monophosphate), the substrates of both ADSL and ATIC respectively (Figure 1), were identified as two major regulator metabolites in yeast [12,13,14]. This suggests that the disease caused by the enzymatic defect could mostly be due to a toxic accumulation of the enzyme-substrates and/or derivatives, rather than by the block in the de novo pathway.
For ADSL, the situation is even more complex as this enzyme acts at two different steps of AMP synthesis and its defect leads to accumulation of its two substrates, SZMP and SAMP (Succinyl AMP). Both metabolites are suspected to contribute to the etiology of the disease, and their relative abundance was proposed to be relevant for symptom severity [37,38], although this assumption is still debated [39,40]. Yeast genetics (our unpublished results) revealed that the ADSL knock-out mutants were genetically unstable and tended to pick-up suppressor mutations upstream in the de novo pathway that would block substrate accumulation. Indeed, in yeast, under conditions where the pathway is constitutively turned-on, SZMP and/or SAICAR (Succinyl Amino Imidazole Carboxamide Ribonucleoside) toxicity clearly correlated to the strength of the allele i.e., to the level of residual ADSL activity [14]. The reasons for SZMP/SAICAR toxicity are not elucidated, but could be related to the regulatory roles of this small molecule. In yeast, SZMP promotes an interaction between two transcription factors, Bas1 and Pho2, and thereby stimulates transcription of the purine regulon [12], while in humans, SZMP specifically stimulates PKM2 in cancer cells [11].
ATIC deficiency is a very rare disease that leads to a massive accumulation of AICAR monophosphate (ZMP) [25]. In yeast, ATIC mutants (the ade17 mutant and ade16 ade17 double mutant) massively accumulate ZMP but also the other nucleosides (AICAR (Amino Imidazole Carboxamide ribonucleoside), SAICAR and Succinyl-adenosine) and nucleotide monophosphate derivatives (SZMP and SAMP). We have shown that an enzymatic inhibition and/or reversion of ADSL (Ade13) by ZMP are responsible for the accumulation of these succinyl derivatives in yeast [12,13]. It could be then expected that the accumulation of these derivatives also observed in ATIC patients [25] could be linked to a similar phenomenon. Yeast thus offers genetic and biochemical tools that can help identify some sources of toxicity for those AICAR derivatives. Indeed, by proteomic approaches, we have recently identified yeast proteins that specifically bind ZMP and some of its derivatives (AICAR, SZMP and AMP) [41]. A similar approach was also performed with mammalian proteins (human cells; M. Duperray, M. Moenner and B. Pinson, unpublished results) and revealed 104 human proteins specifically binding the ZMP resin. Among those human proteins, 70 have a yeast ortholog of which one third were previously identified as specific ZMP binders [41]. Molecular studies of the ZMP effect on some of these proteins can provide new insights into the biological defects associated with human ADSL and/or ATIC deficiencies.
Strikingly, AICAR and ZMP also accumulate in other human purine-associated diseases such as in HGPRT deficiency [42,43], and this was also observed in hpt1 mutant yeast cells [44], suggesting once again a high functional conservation of the metabolic balances in the two organisms. Importantly, a HGPRT deficiency in yeast was synthetically lethal with ZMP accumulation [45], suggesting that the small molecule could contribute to the etiology of the disease. Interestingly, our genetic analyses of yeast revealed mutations in GMP kinase (Guk1, Figure 1) that mimicked the HGPRT deficiency phenotypes: hypoxanthine utilization, purine excretion [23] and also ZMP accumulation [44]. Whether a similar phenocopy mechanism exists in human and which could account for some of the “non-HGPRT-dependent HGPRT-like deficiencies”, remains to be investigated. How and to what extend ZMP accumulation contributes to the pathological symptoms associated with HGPRT-deficiency remains to be established, but this hypothesis has been raised in the past [46].
Interestingly, in yeast, while both ZMP and SZMP are toxic [14], ZMP appeared to be more toxic than SZMP since synthesis of SZMP from ZMP increased AICAR-resistance, while on the other hand blocking synthesis of SZMP from ZMP increased AICAR sensitivity [41]. Beside its potential implication in purine metabolic diseases as a toxic metabolite, AICAR is used as an antiproliferative prodrug. Indeed, AICAR is toxic for tumor cells of multiple origins [47] and in particular to aneuploid cells [48,49] thus raising interesting perspectives as an anticancer molecule. Importantly, it was well tolerated in phase I/II clinical trials [50] and has proved efficient in several different xenograft assays [48,51,52]. Yeast genetics has been used to identify the sources of sensitivity and resistance to AICAR, including uptake [53], carbon utilization [45], nuclear import [41,54] and the ubiquitin pathway [55]. Importantly, based on the results obtained in these yeast studies, we found that AICAR toxicity was increased in the knock-down of human genes (RNF40, ASH2L, MLL2) corresponding to the yeast mutations (bre1, set1) [54]. Hence yeast, besides being a model for purine metabolic diseases, can also be used to reveal genetic backgrounds specifically affected by drugs, including toxic nucleoside analogs such as AICAR [56] or purine metabolism inhibitors such as mycophenolic acid [57], an IMPDH inhibitor which is used as an immunosuppressant in clinics.
4. Yeast as a Tool to Functionally Validate Orthologous Genes from Other Model Organisms
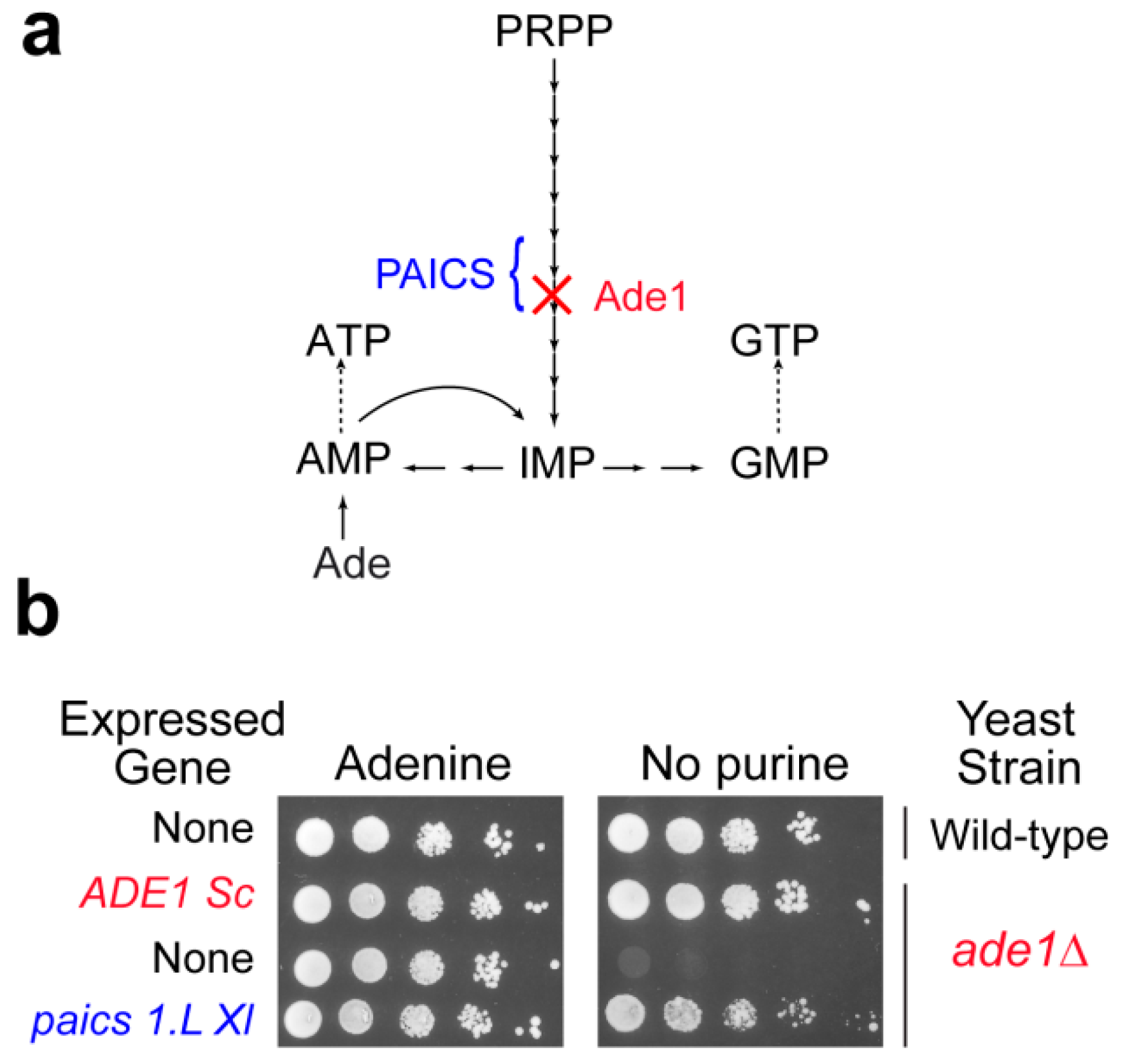
Though yeast studies can be very informative about molecular mechanisms associated with purine metabolism diseases, animal models are required to study tissue specific or developmental aspects that are the most likely determinant in the etiology of purine metabolism diseases. While mice have been used as a model animal, for example to study HGPRT deficiency [58], new models are emerging that are more prone to developmental studies due to external development such as the zebra fish [59] or Xenopus laevis (our unpublished work), or more open to the possibility of running genetic experiments (nematode, [60]). Although the genomes of these organisms are fully sequenced, very little is known about their purine metabolism. As a first step it is essential to identify the purine metabolism coding genes (classically by homology search) and most importantly to validate them functionally. This functional validation can be done by complementation of the corresponding knock-out mutants in yeast. Our unpublished work on X. laevis and on the nematode Caenorhabditis elegans allowed such a functional validation for most of the candidate purine metabolism genes via their expression in yeast. As an illustration, the paics1.L X. laevis gene restored growth of an ade1 knock-out yeast mutant in the absence of external purine (Figure 2 and our unpublished results) or for C. elegans gene candidates [60]. Yeast can also be used for in vivo studies of the effects of mutations in human genes, as was done for example for APRT [61]. Hence, yeast not only proved useful as a model by itself but also to help set up new models more disposed to addressing specific questions such as tissue specificity or developmental aspects. Taking advantage of the complementarity of various models has been a fruitful strategy for tackling highly integrated biological questions, and for providing some understanding about complex human diseases. In this respect, yeast has very efficiently played its part, and will certainly continue to help us address challenging questions in the future.
5. Conclusions
It is striking that the purine de novo pathway is highly conserved between yeast and human (although some enzymes are much more divergent than others), while the salvage pathway shows much more differences between these species (Figure 1). However, despite these specific enzyme differences, several examples reported here reveal a very strong functional conservation, hence highlighting that similar mechanisms ensure homeostasis in both organisms. This functional conservation operates both at the metabolic molecular level, i.e., purine nucleotide content and balance, but also more widely at the cellular or organism level, as revealed by phenotypical outcomes. Due to this highly functional conservation, yeast, which is prone to genetics, proteomic and metabolic studies, provides an interesting model to elaborate new working hypotheses testable in human cells or animal models. In particular, because of the high redundancy of metabolic pathways, combinations of mutants are often required to reveal phenotypes, and while the construction of double or triple mutants is trivial in yeast, it is still challenging in most eukaryote organisms.
Author Contributions
Writing—original draft: B.P. and B.D.-F.; writing—review and editing: B.P. and B.D.-F.
Funding
This research received no external funding.
Acknowledgments
We thank M. Moenner for critically reading the manuscript and M. Duperray and K. Masse for their help in the complementation test, Figure 2.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A

{kind=link}
{kind=link}
Table A1.
List of the Saccharomyces cerevisiae purine pathway enzymes shown in Figure 1. Enzyme features were obtained from the Saccharomyces Genome Database website [62].
| Protein Name | ORF Name | Enzymatic Activities |
|---|---|---|
| Aah1 | YNL141W | Adenine deaminase |
| Ade1 | YAR015W | Phosphoribosyl aminoimidazole succinocarboxamide synthetase |
| Ade12 | YNL220W | Adenylosuccinate synthase |
| Ade13 | YLR359W | Adenylosuccinate lyase |
| Ade16 | YLR028C | AICAR transformylase and IMP cyclohydrolase |
| Ade17 | YMR120C | AICAR transformylase and IMP cyclohydrolase |
| Ade2 | YOR128C | Phosphoribosylaminoimidazole carboxylase |
| Ade4 | YMR300C | Phosphoribosylpyrophosphate amidotransferase |
| Ade5,7 | YGL234W | Aminoimidazole ribonucleotide synthetase and glycinamide ribotide synthetase |
| Ade6 | YGR061C | Formylglycinamidine-ribonucleotide synthetase |
| Ade8 | YDR408C | Phosphoribosyl-glycinamide transformylase |
| Adk1 | YDR226W | Adenylate kinase |
| Adk2 | YER170W | Adenylate kinase |
| Ado1 | YJR105W | Adenosine kinase |
| Amd1 | YML035C | AMP deaminase |
| Apt1 | YML022W | Adenine phosphoribosyltransferase |
| Gua1 | YMR217W | GMP synthase |
| Gud1 | YDL238C | Guanine deaminase |
| Guk1 | YDR454C | Guanylate kinase |
| Hpt1 | YDR399W | Hypoxanthine-guanine phosphoribosyltransferase |
| Imd2 | YHR216W | Inosine monophosphate dehydrogenase |
| Imd3 | YLR432W | Inosine monophosphate dehydrogenase |
| Imd4 | YML056C | Inosine monophosphate dehydrogenase |
| Isn1 | YOR155C | Inosine monophosphate specific nucleotidase |
| Phm8 | YER037W | GMP, UMP and CMP nucleotidase |
| Pnp1 | YLR209C | Purine nucleoside phosphorylase |
| Prs1 | YKL181W | PRPP synthetase subunit |
| Prs2 | YER099C | PRPP synthetase subunit |
| Prs3 | YHL011C | PRPP synthetase subunit |
| Prs4 | YBL068W | PRPP synthetase subunit |
| Prs5 | YOL061W | PRPP synthetase subunit |
| Rnr1 | YER070W | Large subunit of ribonucleotide-diphosphate reductase |
| Rnr2 | YJL026W | Small subunit of ribonucleotide-diphosphate reductase |
| Rnr3 | YIL066C | Large subunit of ribonucleotide-diphosphate reductase |
| Rnr4 | YGR180C | Small subunit of ribonucleotide-diphosphate reductase |
| Ynk1 | YKL067W | Nucleoside diphosphate kinase |
Table A2.
List of the human purine pathway enzymes shown in Figure 1. Enzyme features were obtained from the NCBI web site [63].
| Protein Name | Gene Location | Enzymatic Activity |
|---|---|---|
| ADA | 20q13.12 | Adenosine deaminase |
| ADK | 10q22 | Adenosine kinase |
| ADSL | 22q13.1 | Adenylosuccinate lyase |
| ADSS | 1q44 | Adenylosuccinate synthase |
| AK1 | 9q34.11 | Adenylate kinase |
| AK2 | 1p35.1 | Adenylate kinase |
| AK3 | 9p24.1 | Adenylate kinase |
| AK4 | 1p31.3 | Adenylate kinase |
| AK5 | 1p31.1 | Adenylate kinase |
| AK6 | 5q13.2 | Adenylate kinase |
| AK7 | 14q32.2 | Adenylate kinase |
| AK8 | 9q34.13 | Adenylate kinase |
| AK9 | 6q21 | Adenylate kinase |
| AMPD1 | 1p13.2 | Adenosine monophosphate deaminase |
| AMPD2 | 1p13.3 | Adenosine monophosphate deaminase |
| AMPD3 | 11p15.4 | Adenosine monophosphate deaminase |
| APRT | 16q24.3 | Adenine phosphoribosyltransferase |
| ATIC | 2q35 | Amino-Imidazole CarboxAmide Ribonucleotide transformylase and IMP cyclohydrolase |
| GAH | 9q21.13 | Guanine deaminase |
| GART | 21q22.11 | Phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase and phosphoribosylaminoimidazole synthetase |
| GMPS | 3q25.31 | Guanine monophosphate synthase |
| GUK1 | 1q42.13 | Guanylate kinase |
| GUK2 | 1q32.1-q42 | Guanylate kinase |
| HPRT1 | Xq26.1 | Hypoxanthine phosphoribosyltransferase |
| IMPDH1 | 7q32.1 | Inosine monophosphate dehydrogenase |
| IMPDH2 | 3p21.31 | Inosine monophosphate dehydrogenase |
| NME1 | 17q21.33 | Nucleoside diphosphate kinase |
| NME2 | 17q21.33 | Nucleoside diphosphate kinase |
| NME3 | 16p13.3 | Nucleoside diphosphate kinase |
| NME4 | 16p13.3 | Nucleoside diphosphate kinase |
| NME5 | 5q31.2 | Nucleoside diphosphate kinase |
| NME6 | 3p21.31 | Nucleoside diphosphate kinase |
| NME7 | 1q24.2 | Nucleoside diphosphate kinase |
| PAICS | 4q12 | Phosphoribosylaminoimidazole carboxylase and phosphoribosylaminoimidazolesuccinocarboxamide synthetase |
| PFAS | 17p13.1 | Phosphoribosylformylglycinamidine synthase |
| PNP | 14q11.2 | Purine nucleoside phosphorylase |
| PPAT | 4q12 | Phosphoribosyl pyrophosphate amidotransferase |
| PRPS1 | Xq22.3 | Phosphoribosyl pyrophosphate synthetase 1 |
| PRPS2 | Xp22.2 | Phosphoribosyl pyrophosphate synthetase 2 |
| RRM1 | 11p15.4 | Ribonucleotide reductase catalytic subunit |
| RRM2 | 2p25.1 | Ribonucleotide reductase regulatory subunit |
| RRM2B | 8q22.3 | Ribonucleotide reductase regulatory subunit |
| XDH | 2p23.1 | Xanthine dehydrogenase/oxidase |
Table A3.
Comparison of the yeast and human purine pathway enzymes depicted in Figure 1. Identity and similarity scores were obtained from the ProteomeTM platform [64]. For each enzymatic activity, only the best score obtained between yeast and a human ortholog was shown. Protein coverage refers to the fraction (%) of the entire protein (the smallest of the two orthologs) used in the alignment for determination of identity and similarity scores.
Table A3.
Comparison of the yeast and human purine pathway enzymes depicted in Figure 1. Identity and similarity scores were obtained from the ProteomeTM platform [64]. For each enzymatic activity, only the best score obtained between yeast and a human ortholog was shown. Protein coverage refers to the fraction (%) of the entire protein (the smallest of the two orthologs) used in the alignment for determination of identity and similarity scores.
| Yeast Protein | Human Protein | Identity (%) | Similarity (%) | Protein Coverage (%) |
|---|---|---|---|---|
| Rnr2 | RMM2 | 69 | 81 | 90 |
| Rnr3 | RMM1 | 66 | 84 | 87 |
| Ade13 | ADSL | 64 | 79 | 97 |
| Prs3 | PRPS2 | 61 | 78 | 79 |
| Ynk1 | NME2 | 61 | 80 | 97 |
| Imd2 | IMPD2 | 61 | 78 | 98 |
| Prs2 | PRPS1 | 60 | 75 | 78 |
| Ade16 | ATIC | 60 | 75 | 99 |
| Ade17 | ATIC | 60 | 75 | 99 |
| Adk1 | AK2 | 58 | 73 | 98 |
| Ade12 | ADSS | 52 | 68 | 99 |
| Pnp1 | PNP | 51 | 68 | 85 |
| Guk1 | GUK1 | 51 | 70 | 99 |
| Amd1 | AMPD2 | 50 | 66 | 74 |
| Ade5,7 | GART | 45 | 64 | 99 |
| Apt1 | APRT | 44 | 64 | 96 |
| Gud1 | GAH | 42 | 49 | 99 |
| Ado1 | ADK1 | 36 | 57 | 99 |
| Gua1 | GMPS | 36 | 54 | 100 |
| Ade4 | ATASE/PPAT | 34 | 51 | 92 |
| Ade6 | PFAS | 33 | 49 | 99 |
| Hpt1 | HGPRT | 33 | 46 | 33 |
| Ade8 | GART | 32 | 52 | 92 |
| Ade2 | PAICS | 27 | 45 | 99 |
| Ade1 | PAICS | 22 | 41 | 100 |
References
- Ljungdahl, P.O.; Daignan-Fornier, B. Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae. Genetics 2012, 190, 885–929. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, H.A.; Sabina, R.L.; van den Berghe, G. Metabolic disorders of purine metabolism affecting the nervous system. Handb. Clin. Neurol. 2013, 113, 1827–1836. [Google Scholar] [Green Version]
- Jurecka, A. Inborn errors of purine and pyrimidine metabolism. J. Inherit. Metab. Dis. 2009, 32, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Nyhan, W.L. Disorders of purine and pyrimidine metabolism. Mol. Genet. Metab. 2005, 86, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of purine metabolism: Clinical update and therapies. J. Inherit. Metab. Dis. 2014, 37, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Seegmiller, J.E.; Rosenbloom, F.M.; Kelley, W.N. Enzyme defect associated with a sex-linked human neurological disorder and excessive purine synthesis. Science 1967, 155, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, H.A.; Duley, J.A.; Fairbanks, L.D.; McBride, M.B. When to investigate for purine and pyrimidine disorders. Introduction and review of clinical and laboratory indications. J. Inherit. Metab. Dis. 1997, 20, 214–226. [Google Scholar] [CrossRef]
- Van den Berghe, G.; Vincent, F.; Marie, S. Disorders of purine and pyrimidine metabolism. In Inborn Metabolic Diseases, 4th ed.; Springer: Berlin, Germany, 2006; pp. 433–449. [Google Scholar]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef]
- Keller, K.E.; Doctor, Z.M.; Dwyer, Z.W.; Lee, Y.S. SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol. Cell 2014, 53, 700–709. [Google Scholar] [CrossRef]
- Keller, K.E.; Tan, I.S.; Lee, Y.S. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science 2012, 338, 1069–1072. [Google Scholar] [CrossRef]
- Pinson, B.; Vaur, S.; Sagot, I.; Coulpier, F.; Lemoine, S.; Daignan-Fornier, B. Metabolic intermediates selectively stimulate transcription factor interaction and modulate phosphate and purine pathways. Genes Dev. 2009, 23, 1399–1407. [Google Scholar] [CrossRef] [Green Version]
- Rebora, K.; Desmoucelles, C.; Borne, F.; Pinson, B.; Daignan-Fornier, B. Yeast AMP pathway genes respond to adenine through regulated synthesis of a metabolic intermediate. Mol. Cell Biol. 2001, 21, 7901–7912. [Google Scholar] [CrossRef] [PubMed]
- Rebora, K.; Laloo, B.; Daignan-Fornier, B. Revisiting purine-histidine cross-pathway regulation in Saccharomyces cerevisiae: A central role for a small molecule. Genetics 2005, 170, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Letisse, F.; Absalan, F.; Lu, W.; Kuznetsova, E.; Brown, G. Nucleotide degradation and ribose salvage in yeast. Mol. Syst. Biol. 2013, 9, 665. [Google Scholar] [CrossRef] [PubMed]
- Itoh, R.; Saint-Marc, C.; Chaignepain, S.; Katahira, R.; Schmitter, J.M.; Daignan-Fornier, B. The yeast ISN1 (YOR155c) gene encodes a new type of IMP-specific 5’-nucleotidase. BMC Biochem. 2003, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, V.; Spychala, J. Mammalian 5’-nucleotidases. J. Biol. Chem. 2003, 278, 46195–46198. [Google Scholar] [CrossRef] [PubMed]
- Akeson, A.L.; Wiginton, D.A.; Hutton, J.J. Normal and mutant human adenosine deaminase genes. J. Cell Biochem. 1989, 39, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Ribard, C.; Rochet, M.; Labedan, B.; Daignan-Fornier, B.; Alzari, P.; Scazzocchio, C. Sub-families of alpha/beta barrel enzymes: A new adenine deaminase family. J. Mol. Biol. 2003, 334, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Pinson, B.; Napias, C.; Chevallier, J.; Van den Broek, P.J.; Brethes, D. Characterization of the Saccharomyces cerevisiae cytosine transporter using energizable plasma membrane vesicles. J. Biol. Chem. 1997, 272, 28918–28924. [Google Scholar] [CrossRef]
- Young, J.D.; Yao, S.Y.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Asp. Med. 2013, 34, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Armitt, S.; Woods, R.A. Purine-excreting mutants of Saccharomyces cerevisiae. I. Isolation and genetic analysis. Genet. Res. 1970, 15, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, K.; Konrad, M.; Daignan-Fornier, B. Yeast GMP kinase mutants constitutively express AMP biosynthesis genes by phenocopying a hypoxanthine-guanine phosphoribosyltransferase defect. Genetics 2000, 156, 953–961. [Google Scholar] [PubMed]
- Jaeken, J.; van den Berghe, G. An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids. Lancet 1984, 2, 1058–1061. [Google Scholar] [PubMed]
- Marie, S.; Heron, B.; Bitoun, P.; Timmerman, T.; Van Den Berghe, G.; Vincent, M.F. AICA-ribosiduria: A novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am. J. Hum. Genet. 2004, 74, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.A.; Farrar, G.J.; Kenna, P.; Humphries, M.M.; Sheils, D.M.; Kumar-Singh, R. Localization of an autosomal dominant retinitis pigmentosa gene to chromosome 7q. Nat. Genet. 1993, 4, 54–58. [Google Scholar] [CrossRef]
- Bowne, S.J.; Sullivan, L.S.; Mortimer, S.E.; Hedstrom, L.; Zhu, J.; Spellicy, C.J. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B.; Hansson, O.; Liden, S.; Nilsson, K. Familial ataxic diplegia with deficient cellular immunity. A new clinical entity. Acta Paediatr. Scand. 1970, 59, 545–550. [Google Scholar] [CrossRef] [PubMed]
- De Brouwer, A.P.; Williams, K.L.; Duley, J.A.; van Kuilenburg, A.B.; Nabuurs, S.B.; Egmont-Petersen, M. Arts syndrome is caused by loss-of-function mutations in PRPS1. Am. J. Hum. Genet. 2007, 81, 507–518. [Google Scholar] [CrossRef]
- Park, J.; Hyun, Y.S.; Kim, Y.J.; Nam, S.H.; Kim, S.H.; Hong, Y.B. Exome Sequencing Reveals a Novel PRPS1 Mutation in a Family with CMTX5 without Optic Atrophy. J. Clin. Neurol. 2013, 9, 283–288. [Google Scholar] [CrossRef]
- Synofzik, M.; Muller vom Hagen, J.; Haack, T.B.; Wilhelm, C.; Lindig, T.; Beck-Wodl, S. X-linked Charcot-Marie-Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: Evidence from a family with a novel PRPS1 mutation. Orphanet J. Rare Dis. 2014, 9, 24. [Google Scholar] [CrossRef]
- Becker, M.A.; Puig, J.G.; Mateos, F.A.; Jimenez, M.L.; Kim, M.; Simmonds, H.A. Inherited superactivity of phosphoribosylpyrophosphate synthetase: Association of uric acid overproduction and sensorineural deafness. Am. J. Med. 1988, 85, 383–390. [Google Scholar] [CrossRef]
- Saint-Marc, C.; Pinson, B.; Coulpier, F.; Jourdren, L.; Lisova, O.; Daignan-Fornier, B. Phenotypic consequences of purine nucleotide imbalance in Saccharomyces cerevisiae. Genetics 2009, 183, 529–538. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Schroth, J.; Cai, N.; Vaux, K.; McCloskey, D. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 2013, 154, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, M.P.; Middlemiss, P.J.; Gysbers, J.W.; Andrew, C.; Herman, M.A.; Reed, J.K. Trophic effects of purines in neurons and glial cells. Prog. Neurobiol. 1999, 59, 663–690. [Google Scholar] [CrossRef]
- Jurecka, A.; Zikanova, M.; Kmoch, S.; Tylki-Szymanska, A. Adenylosuccinate lyase deficiency. J. Inherit. Metab. Dis. 2015, 38, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Wadman, S.K.; Duran, M.; van Sprang, F.J.; Beemer, F.A.; Holl, R.A. Adenylosuccinase deficiency: An inborn error of purine nucleotide synthesis. Eur. J. Pediatr. 1988, 148, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Race, V.; Marie, S.; Vincent, M.F.; Van den Berghe, G. Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum. Mol. Genet. 2000, 9, 2159–2165. [Google Scholar] [CrossRef] [Green Version]
- Jurecka, A.; Zikanova, M.; Tylki-Szymanska, A.; Krijt, J.; Bogdanska, A.; Gradowska, W. Clinical, biochemical and molecular findings in seven Polish patients with adenylosuccinate lyase deficiency. Mol. Genet. Metab. 2008, 94, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Zikanova, M.; Skopova, V.; Hnizda, A.; Krijt, J.; Kmoch, S. Biochemical and structural analysis of 14 mutant adsl enzyme complexes and correlation to phenotypic heterogeneity of adenylosuccinate lyase deficiency. Hum. Mutat. 2010, 31, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Douillet, D.C.; Pinson, B.; Ceschin, J.; Hurlimann, H.C.; Saint-Marc, C.; Laporte, D. Metabolomics and proteomics identify the toxic form and the associated cellular binding targets of the anti-proliferative drug AICAR. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Picot, I.; Le Dantec, A.; Brassier, A.; Jais, J.P.; Ledroit, M.; Cahu, J. New biomarkers for early diagnosis of Lesch-Nyhan disease revealed by metabolic analysis on a large cohort of patients. Orphanet J. Rare Dis. 2015, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Sidi, Y.; Mitchell, B.S. Z-nucleotide accumulation in erythrocytes from Lesch-Nyhan patients. J. Clin. Investig. 1985, 76, 2416–2419. [Google Scholar] [CrossRef] [PubMed]
- Hurlimann, H.C.; Laloo, B.; Simon-Kayser, B.; Saint-Marc, C.; Coulpier, F.; Lemoine, S. Physiological and toxic effects of purine intermediate 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR) in yeast. J. Biol. Chem. 2011, 286, 30994–31002. [Google Scholar] [CrossRef] [PubMed]
- Ceschin, J.; Hurlimann, H.C.; Saint-Marc, C.; Albrecht, D.; Violo, T.; Moenner, M. Disruption of Nucleotide Homeostasis by the Antiproliferative Drug 5-Aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside Monophosphate (AICAR). J. Biol. Chem. 2015, 290, 23947–23959. [Google Scholar] [CrossRef]
- Lopez, J.M. Is ZMP the toxic metabolite in Lesch-Nyhan disease? Med. Hypotheses 2008, 71, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Rattan, R.; Giri, S.; Singh, A.K.; Singh, I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J. Biol. Chem. 2005, 280, 39582–39593. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Williams, B.R.; Siegel, J.J.; Amon, A. Identification of aneuploidy-selective antiproliferation compounds. Cell 2011, 144, 499–512. [Google Scholar] [CrossRef]
- Ly, P.; Kim, S.B.; Kaisani, A.A.; Marian, G.; Wright, W.E.; Shay, J.W. Aneuploid human colonic epithelial cells are sensitive to AICAR-induced growth inhibition through EGFR degradation. Oncogene 2013, 32, 3139–3146. [Google Scholar] [CrossRef] [PubMed]
- Van Den Neste, E.; Cazin, B.; Janssens, A.; Gonzalez-Barca, E.; Terol, M.J.; Levy, V. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): A multicenter phase I/II study. Cancer Chemother. Pharmacol. 2013, 71, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937. [Google Scholar] [CrossRef] [Green Version]
- Theodoropoulou, S.; Brodowska, K.; Kayama, M.; Morizane, Y.; Miller, J.W.; Gragoudas, E.S. Aminoimidazole carboxamide ribonucleotide (AICAR) inhibits the growth of retinoblastoma in vivo by decreasing angiogenesis and inducing apoptosis. PLoS ONE 2013, 8, e52852. [Google Scholar] [CrossRef] [PubMed]
- Ceschin, J.; Saint-Marc, C.; Laporte, J.; Labriet, A.; Philippe, C.; Moenner, M. Identification of yeast and human 5-aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside (AICAr) transporters. J. Biol. Chem. 2014, 289, 16844–16854. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, D.; Ceschin, J.; Dompierre, J.; Gueniot, F.; Pinson, B.; Daignan-Fornier, B. Chemo-Genetic Interactions Between Histone Modification and the Antiproliferation Drug AICAR Are Conserved in Yeast and Humans. Genetics 2016, 204, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, D.; Hurlimann, H.C.; Ceschin, J.; Saint-Marc, C.; Pinson, B.; Daignan-Fornier, B. Multiple chemo-genetic interactions between a toxic metabolite and the ubiquitin pathway in yeast. Curr. Genet. 2018, 64, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Daignan-Fornier, B.; Pinson, B. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranosyl 5’-Monophosphate (AICAR), a Highly Conserved Purine Intermediate with Multiple Effects. Metabolites 2012, 2, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Desmoucelles, C.; Pinson, B.; Saint-Marc, C.; Daignan-Fornier, B. Screening the yeast “disruptome” for mutants affecting resistance to the immunosuppressive drug, mycophenolic acid. J. Biol. Chem. 2002, 277, 27036–27044. [Google Scholar] [CrossRef]
- Kuehn, M.R.; et al. A potential animal model for Lesch-Nyhan syndrome through introduction of HPRT mutations into mice. Nature 1987, 326, 295–298. [Google Scholar] [CrossRef]
- Ng, A.; Uribe, R.A.; Yieh, L.; Nuckels, R.; Gross, J.M. Zebrafish mutations in gart and paics identify crucial roles for de novo purine synthesis in vertebrate pigmentation and ocular development. Development 2009, 136, 2601–2611. [Google Scholar] [CrossRef] [Green Version]
- Marsac, R.; Pinson, B.; Saint-Marc, C.; Olmedo, M.; Artal-Sanz, M.; Daignan-Fornier, B.; Gomes, J.-E. Adenylosuccinate Lyase activity in the Purine recycling pathway is essential for developmental timing, germline maintenance and muscle integrity in C. elegans. bioRxiv 2018. [Google Scholar] [CrossRef]
- Huyet, J.; Ozeir, M.; Burgevin, M.C.; Pinson, B.; Chesney, F.; Remy, J.M. Structural Insights into the Forward and Reverse Enzymatic Reactions in Human Adenine Phosphoribosyltransferase. Cell Chem. Biol. 2018, 25, 666–676. [Google Scholar] [CrossRef]
- The Saccharomyces Genome Database. Available online: https://www.yeastgenome.org (accessed on 16 January 2019).
- NCBI. Available online: https://www.ncbi.nlm.nih.gov (accessed on 16 January 2019).
- ProteomeTM Platform. Available online: https://portal.genexplain.com (accessed on 16 January 2019).
Figure 1.
Schematic representation of the human and Saccharomyces cerevisiae purine biosynthesis pathways. Features of human and yeast enzymes are listed in Table A1, Table A2 and are shown in black and pink, respectively. For human enzymes, the numerous isoforms detected for some enzymes are not presented. Ortholog identity scores were determined as described in supplementary Table A3. Grey boxes correspond to enzymes with no or unidentified orthologs between yeast and human. “ATP production” in both yeast and human cells corresponds to the different ATP production pathways such as glycolysis and the respiratory chain/ATP synthase complexes. “Nucl.” stands for the numerous human nucleotidases, such as for example the NT5 family. Abbreviations: IMP: Inosine monophosphate; PRPP: Phosphorybosyl pyrophosphate; SAMP: Succinyl-AMP; SZMP: Succinyl Amino Imidazole Carboxamide Ribonucleotide monophosphate; XMP: Xanthosine monophosphate; ZMP: Amino Imidazole CarboxAmide Ribonucleotide monophosphate.
Figure 1.
Schematic representation of the human and Saccharomyces cerevisiae purine biosynthesis pathways. Features of human and yeast enzymes are listed in Table A1, Table A2 and are shown in black and pink, respectively. For human enzymes, the numerous isoforms detected for some enzymes are not presented. Ortholog identity scores were determined as described in supplementary Table A3. Grey boxes correspond to enzymes with no or unidentified orthologs between yeast and human. “ATP production” in both yeast and human cells corresponds to the different ATP production pathways such as glycolysis and the respiratory chain/ATP synthase complexes. “Nucl.” stands for the numerous human nucleotidases, such as for example the NT5 family. Abbreviations: IMP: Inosine monophosphate; PRPP: Phosphorybosyl pyrophosphate; SAMP: Succinyl-AMP; SZMP: Succinyl Amino Imidazole Carboxamide Ribonucleotide monophosphate; XMP: Xanthosine monophosphate; ZMP: Amino Imidazole CarboxAmide Ribonucleotide monophosphate.

Figure 2.
Functional complementation of the yeast phosphoribosyl aminoimidazole succinocarboxamide synthetase by the Xenopus laevis ortholog paics1.L. (a). Simplified scheme of the yeast purine pathway. (b). Yeast wild-type and knock-out mutant (ade1Δ) strains were either transformed with a plasmid allowing expression of the yeast (ADE1 Sc) or the Xenopus laevis (paics 1.L) amino imidazole succinocarboxamide synthetase encoded gene, or with the empty vector (None). Serial dilutions (1/10) of transformants were dropped on SDCASAW medium to score the ability of the yeast and xenopus genes to complement the ade1 mutant auxotrophy observed in the absence of external purine source. A medium supplemented with adenine was used as a viability control of transformants and images were obtained after 34 h of growth at 37 °C.
Figure 2.
Functional complementation of the yeast phosphoribosyl aminoimidazole succinocarboxamide synthetase by the Xenopus laevis ortholog paics1.L. (a). Simplified scheme of the yeast purine pathway. (b). Yeast wild-type and knock-out mutant (ade1Δ) strains were either transformed with a plasmid allowing expression of the yeast (ADE1 Sc) or the Xenopus laevis (paics 1.L) amino imidazole succinocarboxamide synthetase encoded gene, or with the empty vector (None). Serial dilutions (1/10) of transformants were dropped on SDCASAW medium to score the ability of the yeast and xenopus genes to complement the ade1 mutant auxotrophy observed in the absence of external purine source. A medium supplemented with adenine was used as a viability control of transformants and images were obtained after 34 h of growth at 37 °C.

Table 1.
Pathologies associated with defects in purine synthesis enzymes highly conserved between yeast and humans.
Table 1.
Pathologies associated with defects in purine synthesis enzymes highly conserved between yeast and humans.
| Pathology | Enzymatic Defect | Gene Name (Location) | Phenotype MIM Number | Inheritance | Locus MIM Number | Reference |
|---|---|---|---|---|---|---|
| ADSL deficiency | Loss of function | ADSL (22q13.1) | 103050 | Autosomal recessive | 608222 | [24] |
| AICA-Ribosiduria | Loss of function | ATIC (2q35) | 608688 | Autosomal recessive | 601731 | [25] |
| Retinitis pigmentosa 10 | Loss of function | IMPDH1 (7q32.1) | 180105 | Autosomal dominant | [26] | |
| Leber Congenital Amaurosis 11 | Loss of function | IMPDH1 (7q32.1) | 613837 | ? | 146690 | [27] |
| PNP deficiency | PNP (14q11.2) | 613179 | Autosomal recessive | 164050 | [28] | |
| Arts syndrome | Loss of function | PRPS1 (Xq22.3) | 311835 | X-linked recessive | 311850 | [29] |
| Charcot-Marie-Tooth disease, X-linked recessive, 5 | Loss of function | PRPS1 (Xq22.3) | 311070 | X-linked recessive | 311850 | [30] |
| Deafness, X-linked 1 | Loss of function | PRPS1 (Xq22.3) | 304500 | X-linked | 311850 | [31] |
| Hyperuricemia, PRPS-related | Gain of function | PRPS1 (Xq22.3) | 300661 | X-linked recessive | 311850 | [32] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Daignan-Fornier, B.; Pinson, B. Yeast to Study Human Purine Metabolism Diseases. Cells 2019, 8, 67. https://doi.org/10.3390/cells8010067
AMA Style
Daignan-Fornier B, Pinson B. Yeast to Study Human Purine Metabolism Diseases. Cells. 2019; 8(1):67. https://doi.org/10.3390/cells8010067
Chicago/Turabian StyleDaignan-Fornier, Bertrand, and Benoît Pinson. 2019. "Yeast to Study Human Purine Metabolism Diseases" Cells 8, no. 1: 67. https://doi.org/10.3390/cells8010067
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.