Gap Junction Intercellular Communication Positively Regulates Cisplatin Toxicity by Inducing DNA Damage through Bystander Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
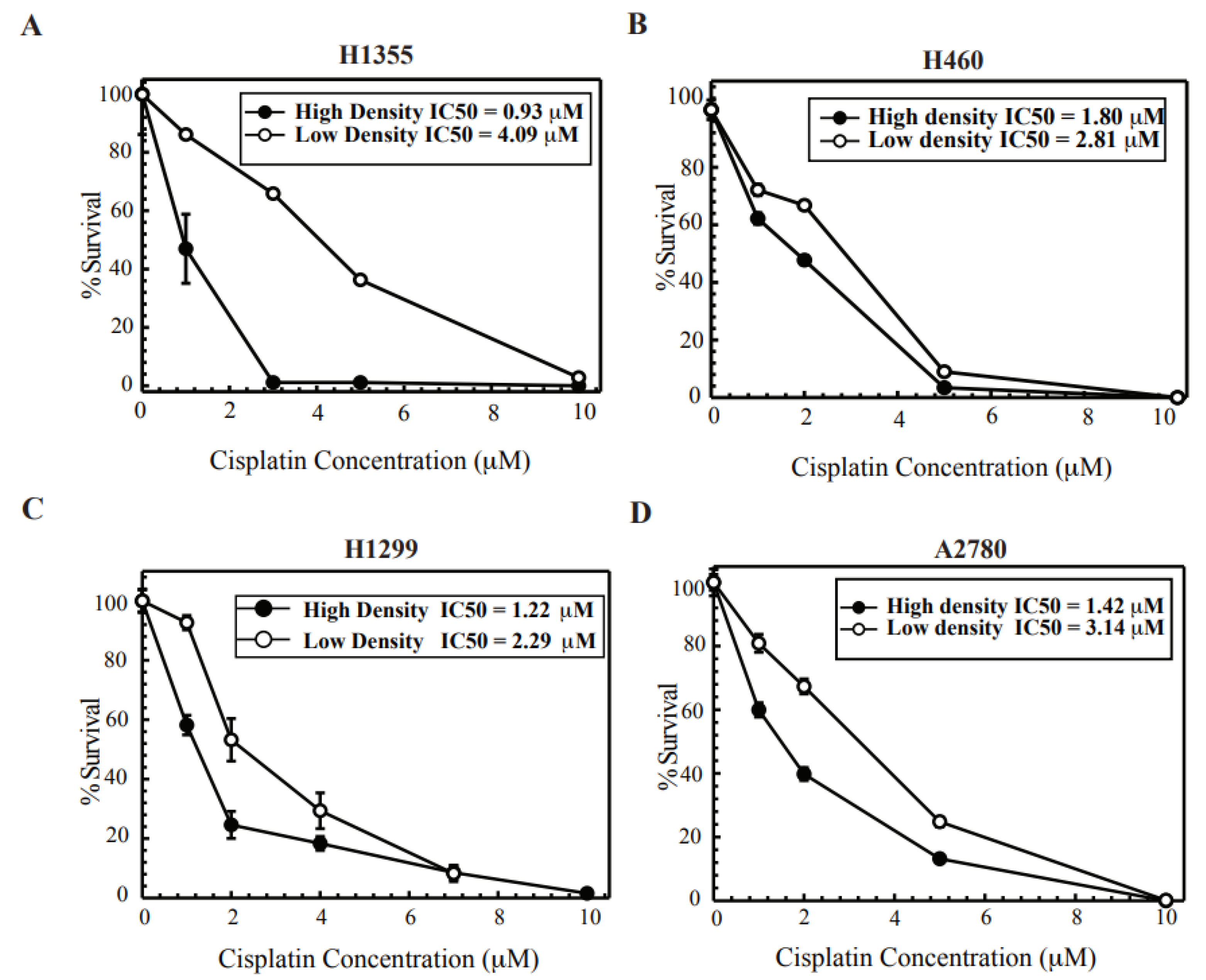
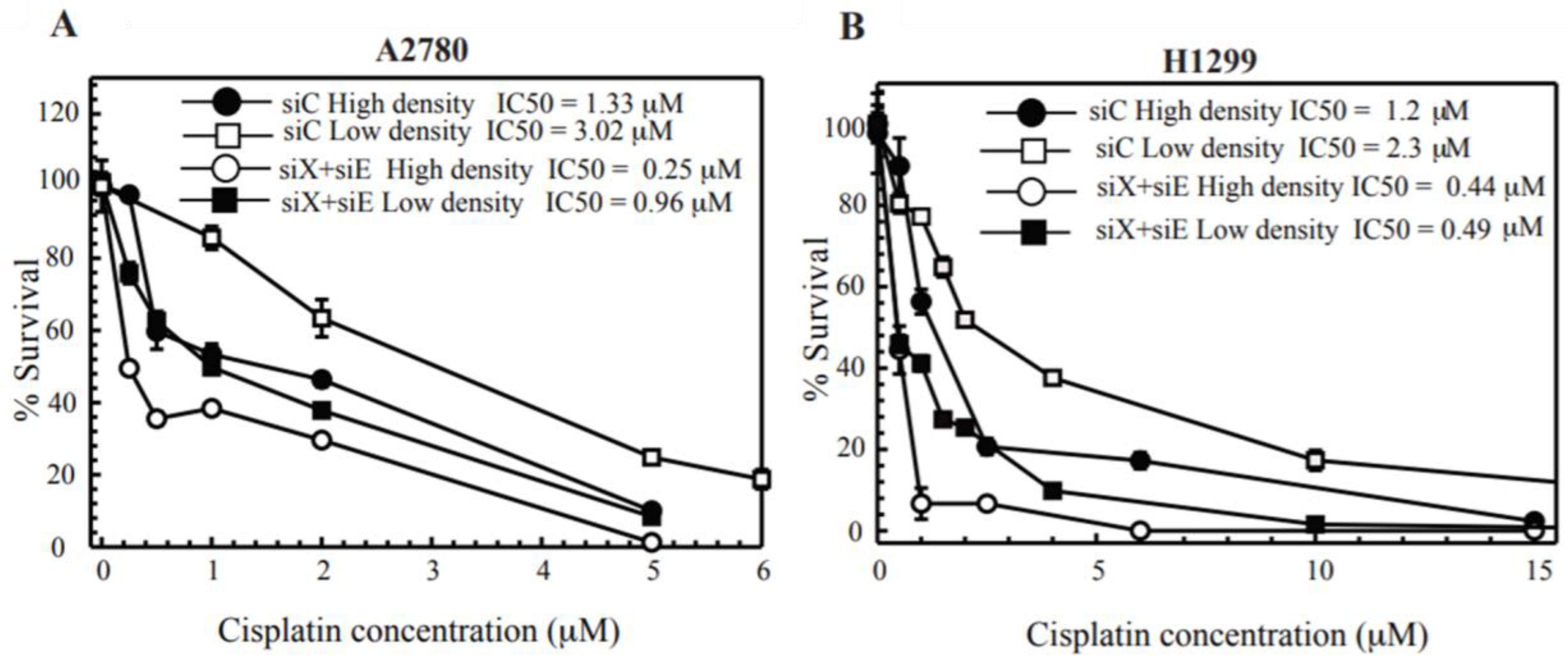
2.1. Cisplatin Treatment at High Density Sensitizes Cancer Cell Lines
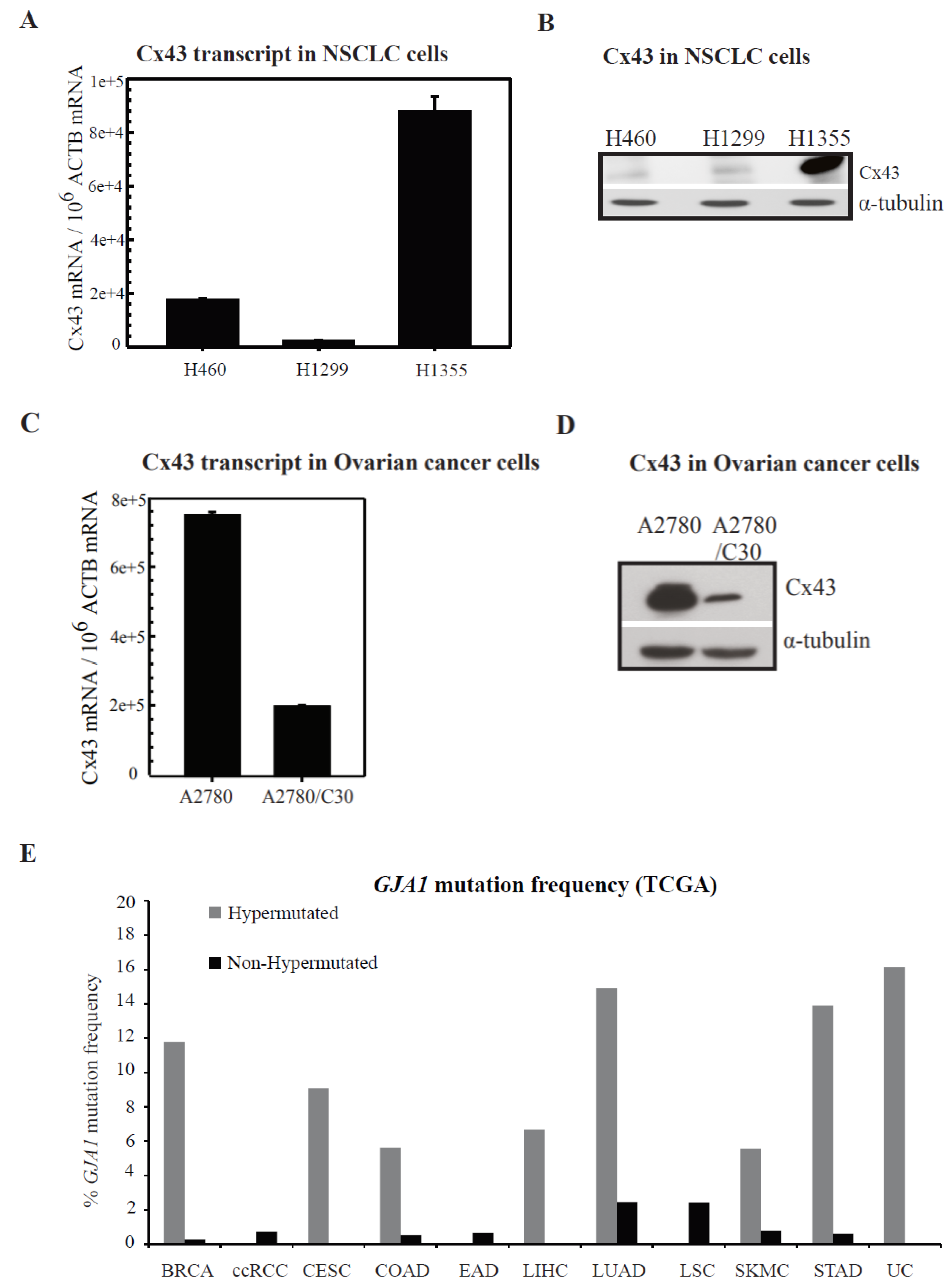
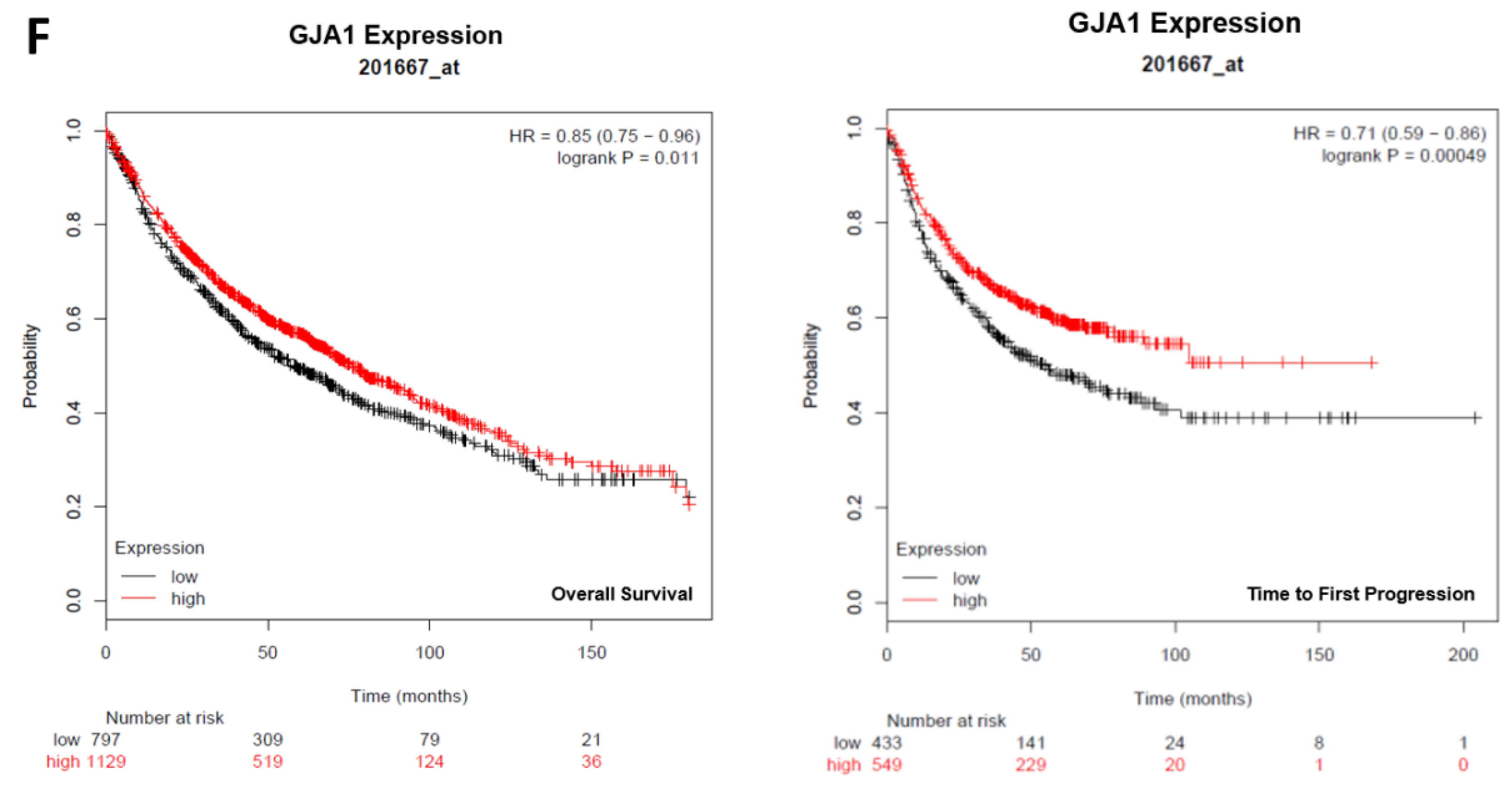
2.2. Connexin Expression in Lung and Ovarian Cancer Cells
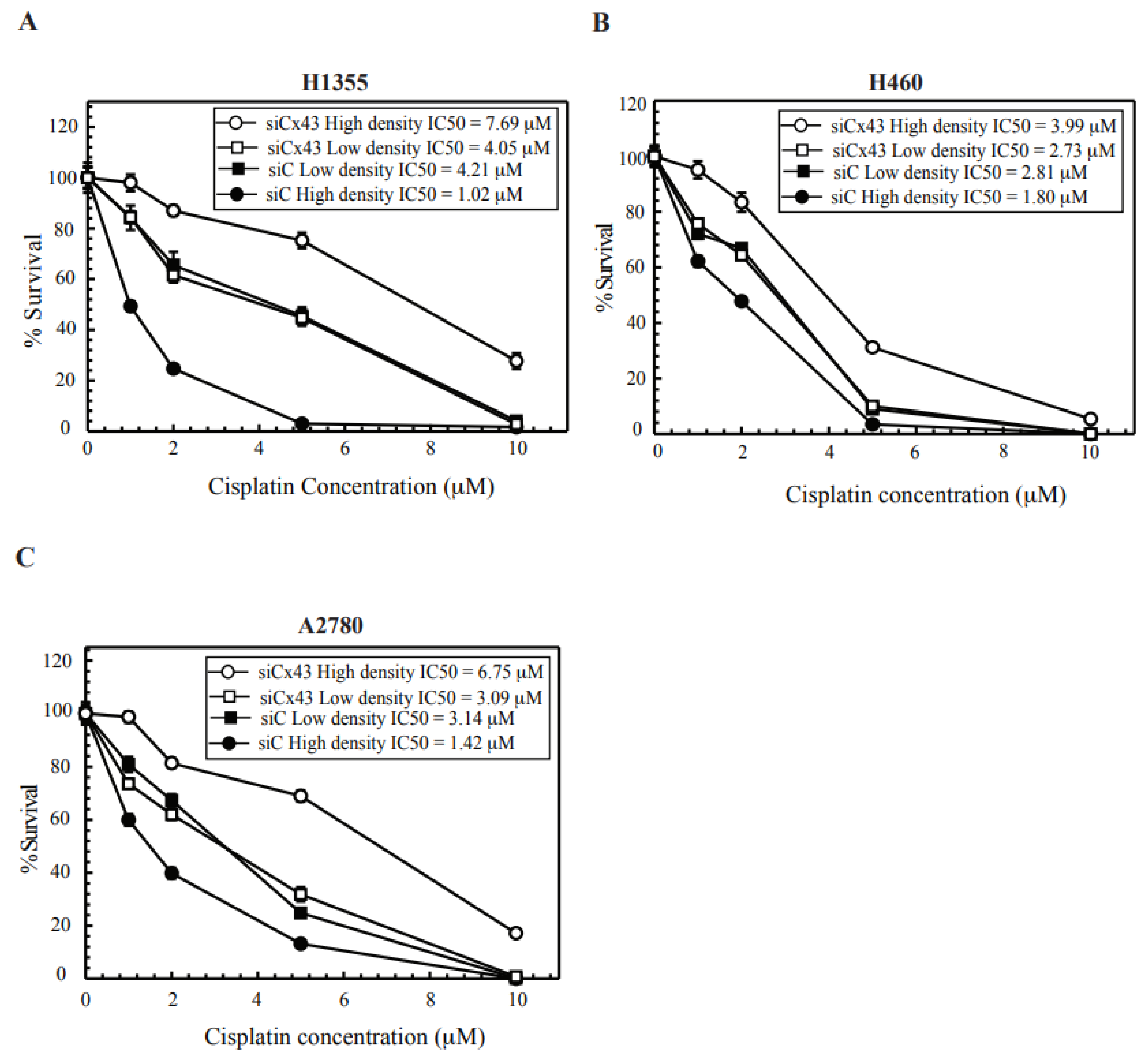
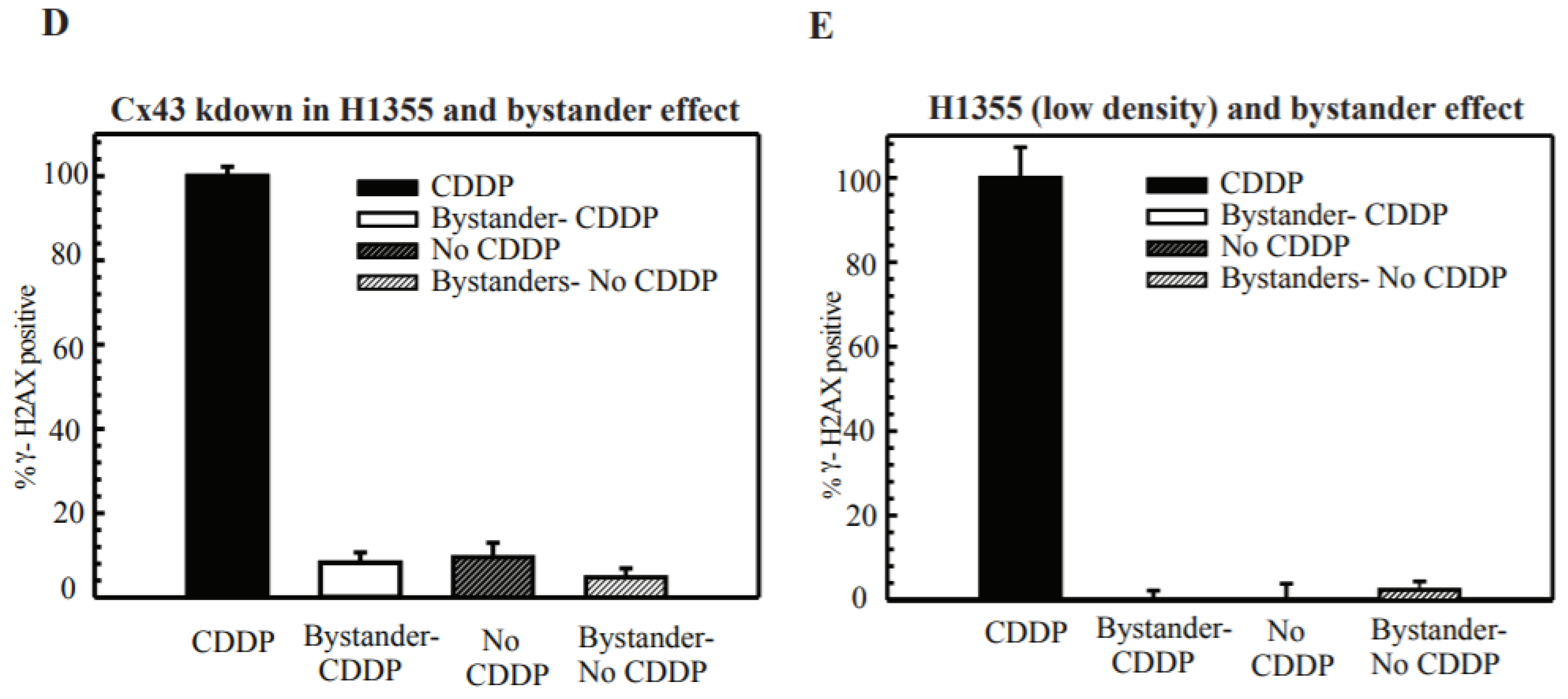
2.3. Cx43 Knockdown Cells Leads to Cisplatin Resistance at High-Density Treatment
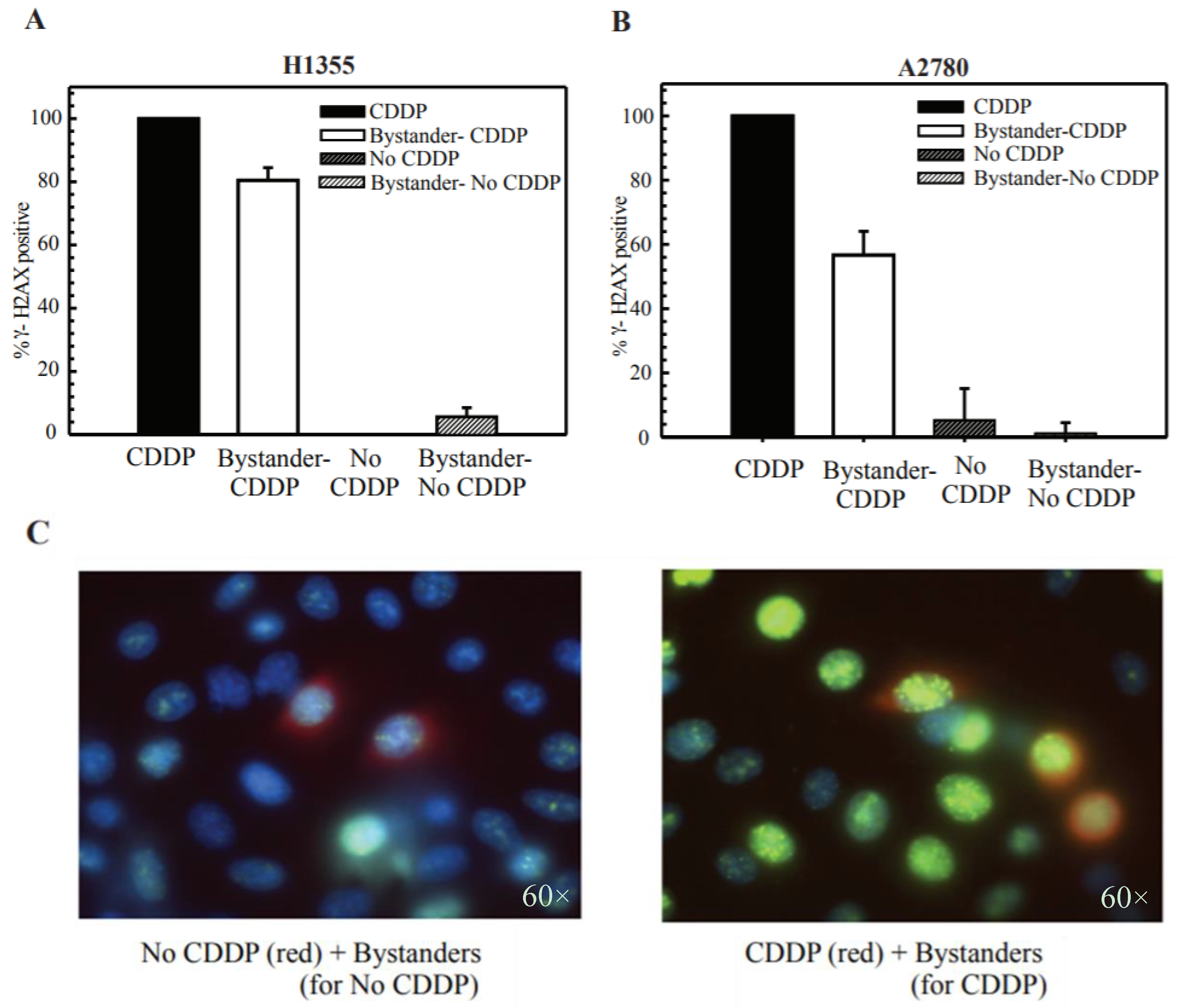
2.4. Cisplatin Treatment Produces DNA DSBs in Bystander Cells
2.5. DSB Production in Bystander Cells Depends on Functional GJIC
2.6. Functional Gap Junction Communication Potentiates Cisplatin Cytotoxicity in DNA Repair Deficient Cells
3. Discussion
4. Materials and Methods
4.1. Colony Survival Assay
4.2. Chemicals
4.3. Cell Culture
4.4. siRNA Sequence and Transfections
4.5. Western Blot
4.6. RNA Isolation, Reverse Transcription and Transcript Abundance
4.7. Lucifer Yellow Dye-Transfer Assay
4.8. Gamma-H2AX Phosphorylation for DNA DSB Measurement
4.9. Immunofluorescence with Cisplatin-DNA Intra-Adduct Specific Antibody
4.10. Mutation Frequency and Survival Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NSCLC | Non-small cell lung cancer |
| GJ | Gap junction |
| GJIC | Gap junction intercellular communication |
| ICL | Interstrand crosslinks |
| DSBs | Double strand breaks |
| Cx43 | Connexin-43 |
| ERCC1 | Excision repair cross-complementation group 1 |
| XPF | Xeroderma pigmentation group F |
| siRNA | Small interfering RNA |
| StaRT–PCR | Standardized reverse transcription–polymerase chain reaction |
| ACTB | β-Actin |
| RIBE | Radiation induced bystander effect |
| BE | Bystander effect |
| SAHA | Suberoylanilide hydroxamic acid |
References
- Cooley, M.E.; Davis, L.; Abrahm, J. Cisplatin: A clinical review. Part II—Nursing assessment and management of side effects of cisplatin. Cancer Nurs. 1994, 17, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Cooley, M.E.; Davis, L.E.; DeStefano, M.; Abrahm, J. Cisplatin: A clinical review. Part I—Current uses of cisplatin and administration guidelines. Cancer Nurs. 1994, 17, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Lippard, S.J. Cisplatin: From DNA damage to cancer chemotherapy. Prog. Nucleic Acid Res. Mol. Biol. 2001, 67, 93–130. [Google Scholar] [PubMed]
- Wernyj, R.P.; Morin, P.J. Molecular mechanisms of platinum resistance: Still searching for the Achilles’ heel. Drug Resist. Updat. 2004, 7, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.; Glazer, P.M. Cell-interdependent cisplatin killing by Ku/DNA-dependent protein kinase signaling transduced through gap junctions. Proc. Natl. Acad Sci. USA 2004, 101, 6134–6139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Li, B.; Liu, H.; Yuan, M.; Yu, M.; Tao, L.; Dong, S.; Tong, X. In vitro inhibited effect of gap junction composed of Cx43 in the invasion and metastasis of testicular cancer resistanced to cisplatin. Biomed. Pharmacother. 2018, 98, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Zhang, Q. Deficiency of gap junction composed of connexin43 contributes to oxaliplatin resistance in colon cancer cells. Oncol. Lett. 2017, 14, 3669–3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Nguyen, T.A. Gap Junction Enhancer Potentiates Cytotoxicity of Cisplatin in Breast Cancer Cells. J. Cancer Sci. Ther. 2012, 4, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rodriguez, L.; Perez-Torras, S.; Carrio, M.; Cascante, A.; Garcia-Ribas, I.; Mazo, A.; Fillat, C. Connexin-26 is a key factor mediating gemcitabine bystander effect. Mol. Cancer Ther. 2011, 10, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Di, X.; Bright, A.T.; Bellott, R.; Gaskins, E.; Robert, J.; Holt, S.; Gewirtz, D.; Elmore, L. A chemotherapy-associated senescence bystander effect in breast cancer cells. Cancer Biol. Ther. 2008, 7, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandre, J.; Hu, Y.; Lu, W.; Pelicano, H.; Huang, P. Novel action of paclitaxel against cancer cells: Bystander effect mediated by reactive oxygen species. Cancer Res. 2007, 67, 3512–3517. [Google Scholar] [CrossRef] [PubMed]
- Blyth, B.J.; Sykes, P.J. Radiation-induced bystander effects: What are they, and how relevant are they to human radiation exposures? Radiat. Res. 2011, 176, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, V.A. Role of nitric oxide in the radiation-induced bystander effect. Redox Biol. 2015, 6, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; De Toledo, S.M.; Little, J.B. Oxidative metabolism, gap junctions and the ionizing radiation-induced bystander effect. Oncogene 2003, 22, 7050–7057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzam, E.I.; De Toledo, S.M.; Little, J.B. Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from alpha-particle irradiated to nonirradiated cells. Proc. Natl. Acad Sci. USA 2001, 98, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Vanhaecke, T.; Papeleu, P.; Snykers, S.; Henkens, T.; Rogiers, V. Connexins and their channels in cell growth and cell death. Cell. Signal. 2006, 18, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Mesnil, M.; Crespin, S.; Avanzo, J.L.; Zaidan-Dagli, M.L. Defective gap junctional intercellular communication in the carcinogenic process. Biochim. Biophys. Acta 2005, 1719, 125–145. [Google Scholar] [CrossRef] [PubMed]
- Kalvelyte, A.; Imbrasaite, A.; Bukauskiene, A.; Verselis, V.K.; Bukauskas, F.F. Connexins and apoptotic transformation. Biochem. Pharmacol. 2003, 66, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Peterson-Roth, E.; Brdlik, C.M.; Glazer, P.M. Src-Induced Cisplatin Resistance Mediated by Cell-to-Cell Communication. Cancer Res. 2009, 69, 3619–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trosko, J.E.; Ruch, R.J. Gap junctions as targets for cancer chemoprevention and chemotherapy. Curr. Drug Targets 2002, 3, 465–482. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, S.; Going, J.J.; D’Arcy, R.; George, W.D. Expression of gap junction proteins connexin 26 and connexin 43 in normal human breast and in breast tumours. J. Pathol. 1998, 184, 37–43. [Google Scholar] [CrossRef]
- El-Fouly, M.H.; Trosko, J.E.; Chang, C.C. Scrape-loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Exp. Cell. Res. 1987, 168, 422–430. [Google Scholar] [CrossRef]
- Hong, X.T.; Wang, Q.; Yang, Y.; Zheng, S.P.; Tong, X.H.; Zhang, S.Z.; Tao, L.; Harris, A.L. Gap junctions propagate opposite effects in normal and tumor testicular cells in response to cisplatin. Cancer Lett. 2012, 317, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Hei, T.K.; Zhou, H.; Ivanov, V.N.; Hong, M.; Lieberman, H.B.; Brenner, D.J.; Amundson, S.A.; Geard, C.R. Mechanism of radiation-induced bystander effects: A unifying model. J. Pharm. Pharmacol. 2008, 60, 943–950. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Tong, X.H.; Wang, L.Z.; Wang, Q.; Ye, H.; Liu, B.; Hong, X.T.; Tao, L.; Harris, A.L. Tramadol and Flurbiprofen Depress the Cytotoxicity of Cisplatin via Their Effects on Gap Junctions. Clin. Cancer Res. 2009, 15, 5803–5810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorgen, P.L.; Trease, A.J.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein(-)Protein Interactions with Connexin 43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, M.V.; Smilenov, L.B.; Hall, E.J.; Panyutin, I.G.; Bonner, W.M.; Sedelnikova, O.A. Ionizing radiation induces DNA double-strand breaks in bystander primary human fibroblasts. Oncogene 2005, 24, 7257–7265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, P.L.; Banath, J.P.; Durand, R.E. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the "comet" assay. Radiat Res. 1990, 122, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Kothandapani, A.; Tillison, K.; Kalman-Maltese, V.; Patrick, S.M. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair (Amst) 2010, 9, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.R.; Ismail-Khan, R.; Bepler, G. Nuclear excision repair-based personalized therapy for non-small cell lung cancer: From hypothesis to reality. Int. J. Biochem. Cell. Biol. 2007, 39, 1318–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, G.R.; Begum, M.; Bepler, G. Setting the stage for tailored chemotherapy in the management of non-small cell lung cancer. Future Oncol. 2008, 4, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Kaye, S.B.; Yap, T.A. PARP inhibitors: The race is on. Br. J. Cancer 2016, 114, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, D.; Carette, D.; Segretain, D.; Gilleron, J.; Pointis, G. Connexin 43 a check-point component of cell proliferation implicated in a wide range of human testis diseases. Cell. Mol. Life Sci. 2013, 70, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A. tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, H.; Krutovskikh, V.; Mesnil, M.; Tanaka, T.; Zaidan-Dagli, M.L.; Omori, Y. Role of connexin (gap junction) genes in cell growth control and carcinogenesis. CR Acad Sci. III 1999, 322, 151–159. [Google Scholar] [CrossRef]
- Klammer, H.; Mladenov, E.; Li, F.; Iliakis, G. Bystander effects as manifestation of intercellular communication of DNA damage and of the cellular oxidative status. Cancer Lett. 2015, 356, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.C.; Xiao, H.L.; Jiang, X.F.; Wang, Q.L.; Li, Y.; Yang, X.J.; Ping, Y.F.; Duan, J.J.; Jiang, J.Y.; Ye, X.Z.; et al. Connexin 43 reverses malignant phenotypes of glioma stem cells by modulating E.-cadherin. Stem Cells 2012, 30, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Stahl, W. Carrots, tomatoes and cocoa: Research on dietary antioxidants in Dusseldorf. Arch. Biochem. Biophys. 2016, 595, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E. The role of stem cells and gap junctions as targets for cancer chemoprevention and chemotherapy. Biomed. Pharmacother. 2005, 59 (Suppl. 2), S326–S331. [Google Scholar] [CrossRef]
- Kelsey, L.; Katoch, P.; Ray, A.; Mitra, S.; Chakraborty, S.; Lin, M.F.; Mehta, P.P. Vitamin D3 regulates the formation and degradation of gap junctions in androgen-responsive human prostate cancer cells. PLoS ONE 2014, 9, e106437. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Hayashi, T.; Tokunou, M.; Nakachi, K.; Trosko, J.E.; Chang, C.C.; Yorioka, N. Suberoylanilide hydroxamic acid enhances gap junctional intercellular communication via acetylation of histone containing connexin 43 gene locus. Cancer Res. 2005, 65, 9771–9778. [Google Scholar] [CrossRef] [PubMed]
- Shishido, S.N.; Nguyen, T.A. Gap junction enhancer increases efficacy of cisplatin to attenuate mammary tumor growth. PLoS ONE 2012, 7, e44963. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.A.; Crawford, E.L.; Warner, K.A.; Elkhairi, F.; Khuder, S.A.; Willey, J.C. ABCC5, ERCC2, XPA and XRCC1 transcript abundance levels correlate with cisplatin chemoresistance in non-small cell lung cancer cell lines. Mol. Cancer 2005, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willey, J.C.; Crawford, E.L.; Jackson, C.M.; Weaver, D.A.; Hoban, J.C.; Khuder, S.A.; DeMuth, J.P. Expression measurement of many genes simultaneously by quantitative RT-PCR using standardized mixtures of competitive templates. Am. J. Respir. Cell. Mol. Biol. 1998, 19, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arora, S.; Heyza, J.R.; Chalfin, E.C.; Ruch, R.J.; Patrick, S.M. Gap Junction Intercellular Communication Positively Regulates Cisplatin Toxicity by Inducing DNA Damage through Bystander Signaling. Cancers 2018, 10, 368. https://doi.org/10.3390/cancers10100368
Arora S, Heyza JR, Chalfin EC, Ruch RJ, Patrick SM. Gap Junction Intercellular Communication Positively Regulates Cisplatin Toxicity by Inducing DNA Damage through Bystander Signaling. Cancers. 2018; 10(10):368. https://doi.org/10.3390/cancers10100368
Chicago/Turabian StyleArora, Sanjeevani, Joshua R. Heyza, Elaine C. Chalfin, Randall J. Ruch, and Steve M. Patrick. 2018. "Gap Junction Intercellular Communication Positively Regulates Cisplatin Toxicity by Inducing DNA Damage through Bystander Signaling" Cancers 10, no. 10: 368. https://doi.org/10.3390/cancers10100368