Targeting MicroRNA-143 Leads to Inhibition of Glioblastoma Tumor Progression

 , ,
, ,
Abstract
:1. Introduction
2. Results
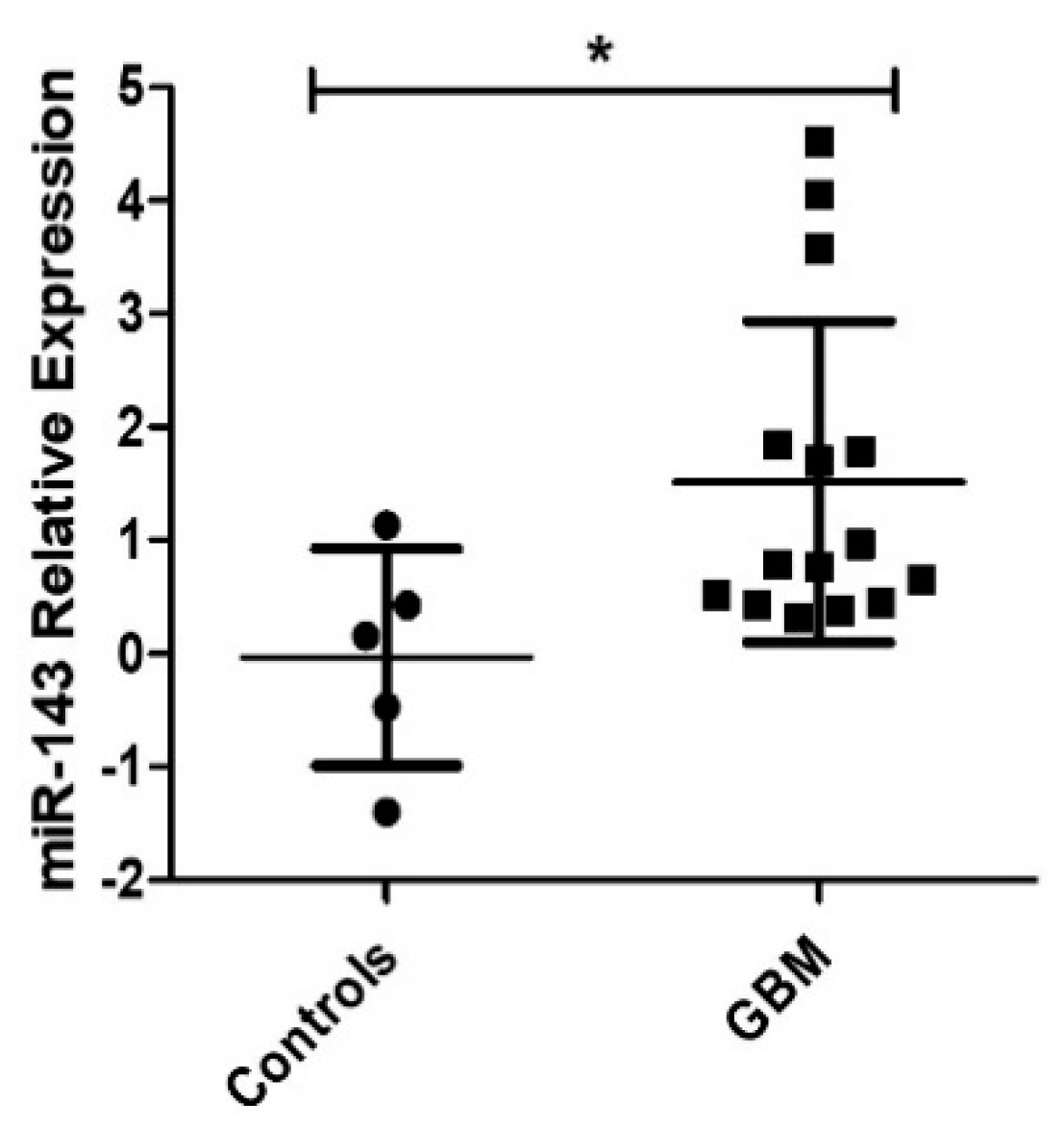
2.1. Expression of MiR-143 in GBM Patients
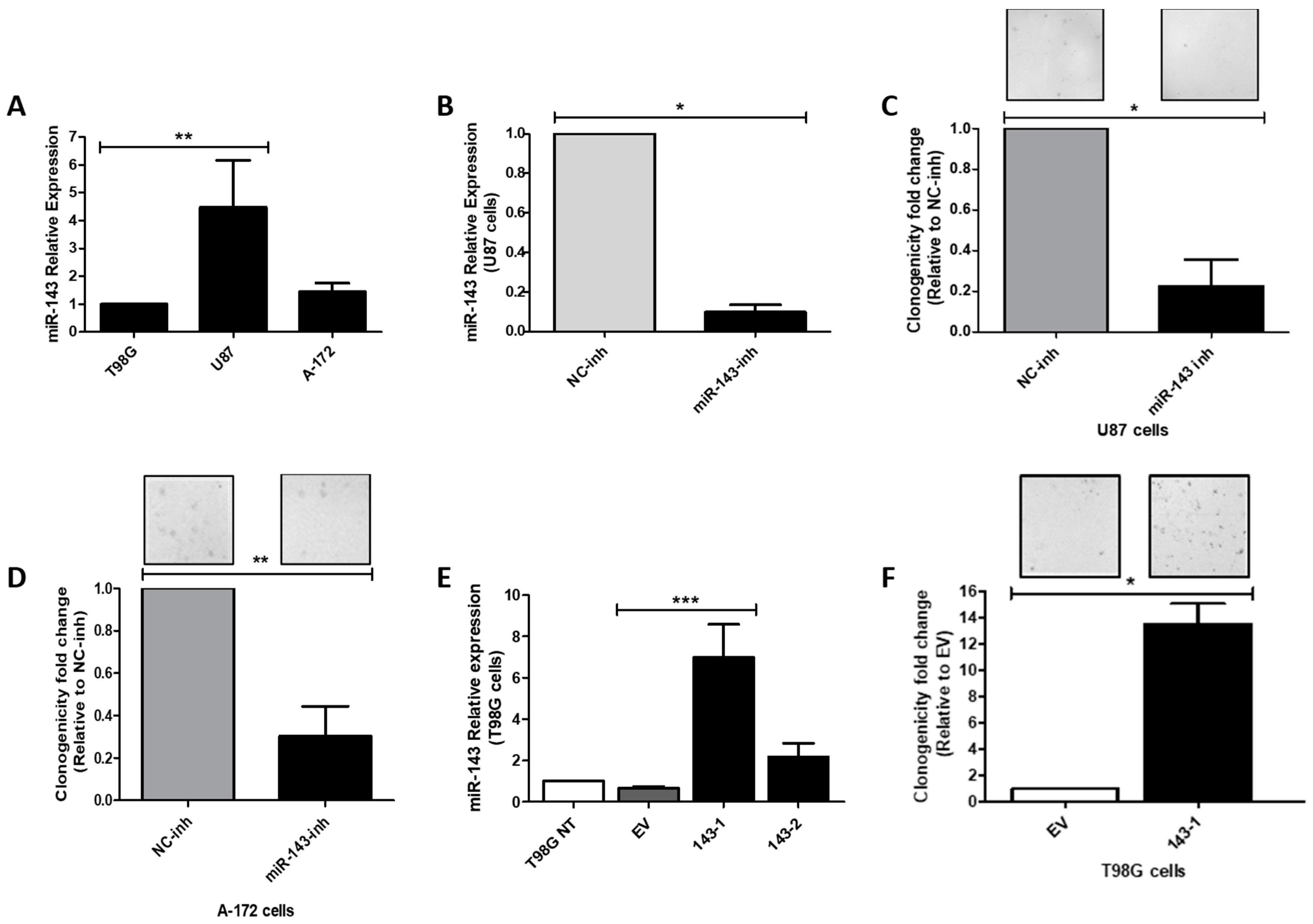
2.2. Effect of MiR-143 Targeting on GBM Cell Proliferation
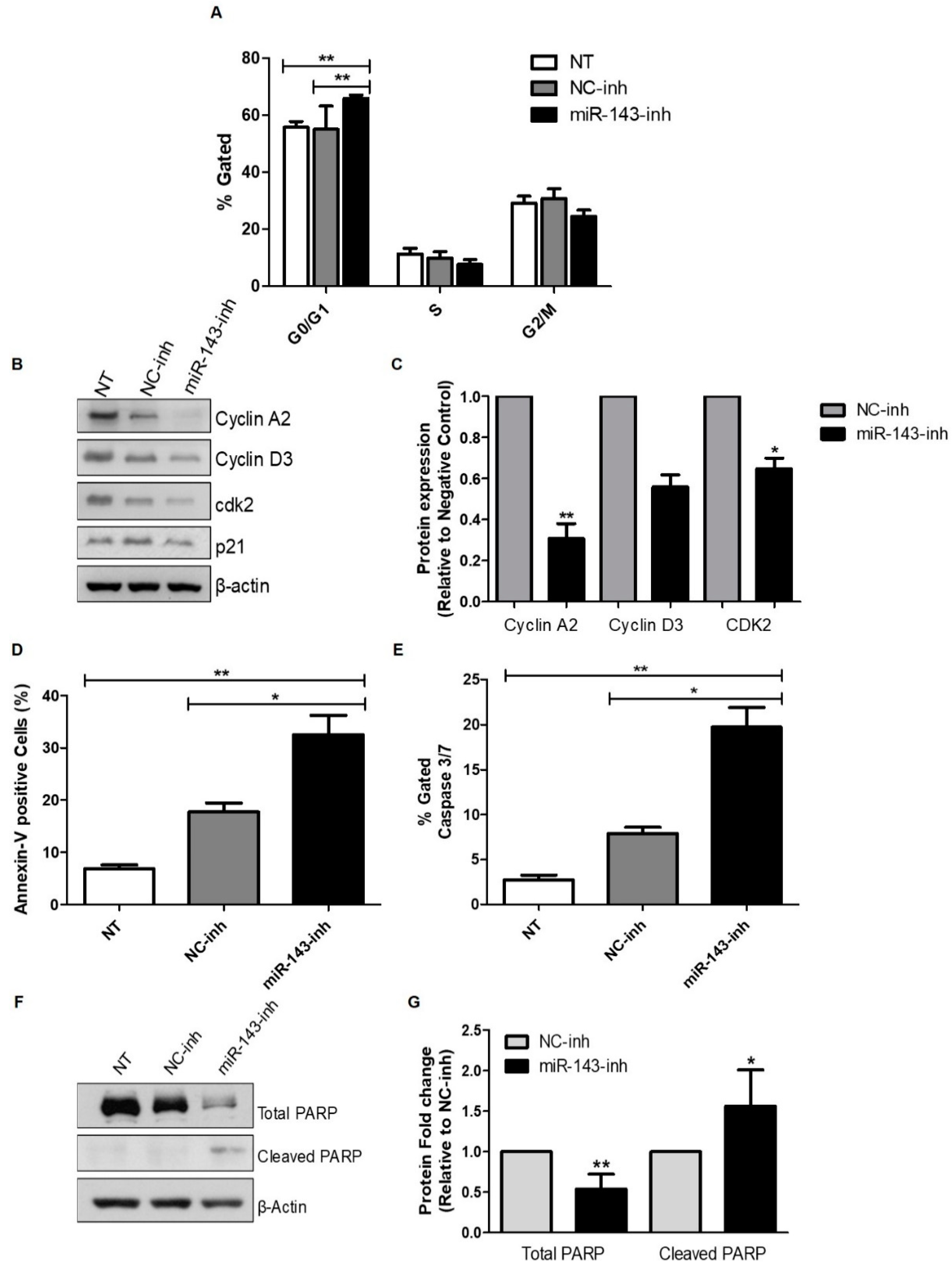
2.3. Effect of MiR-143 Targeting on Cell Cycle Progression and Apoptosis
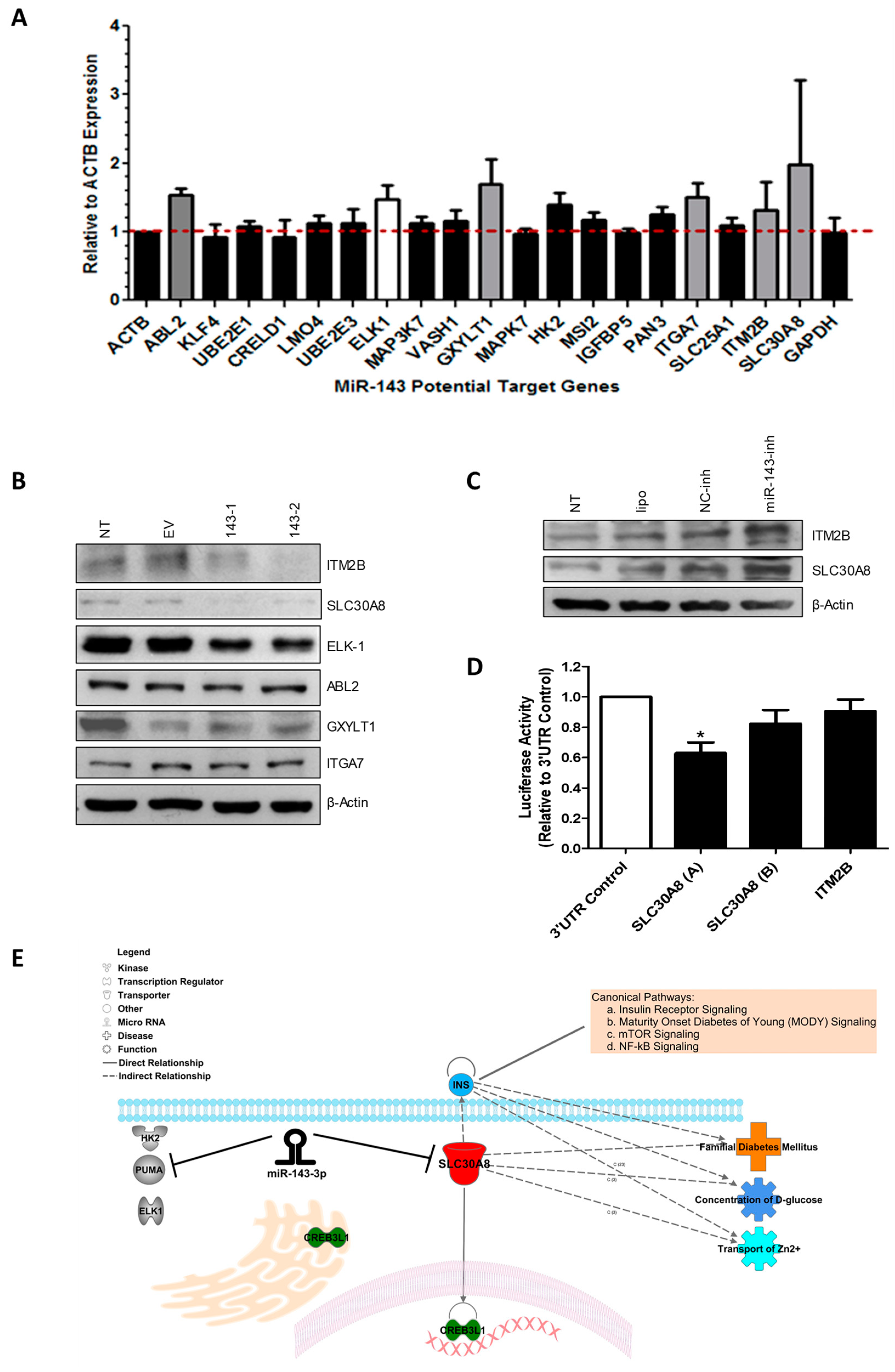
2.4. MiR-143 Target Prediction and Validation
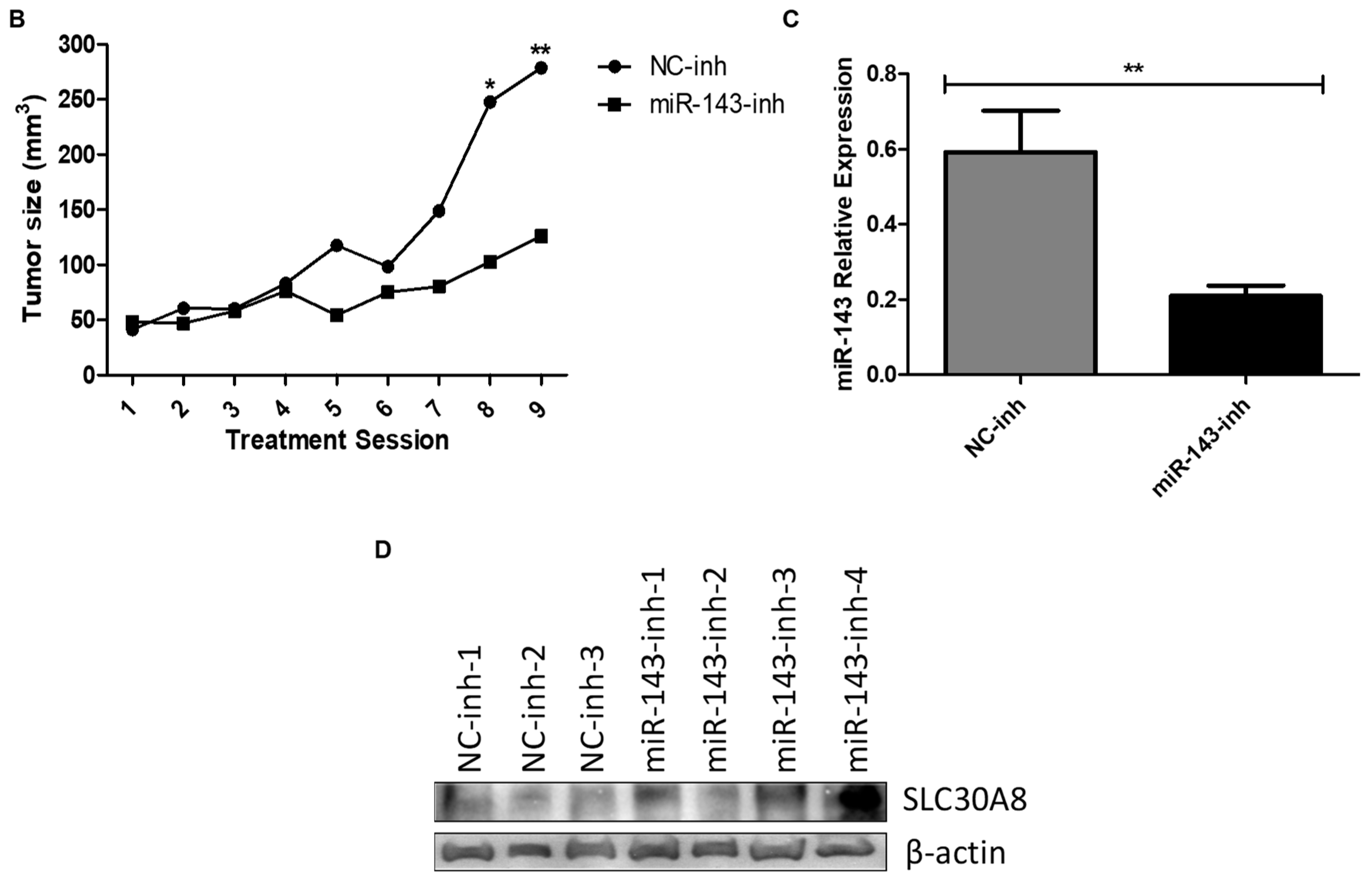
2.5. In Vivo Targeting of MiR-143
3. Discussion
4. Materials and Methods
4.1. Tumor Samples: RNA and Protein Isolation
4.2. Cells and Culture Conditions
4.3. Transient and Stable Transfections
4.4. Colony Formation Assay
4.5. Assessment of Cell Cycle and Apoptosis
4.6. MiRNA Target Prediction and SYBR-Green I RT-PCR for Target Identification
4.7. Western Blot Analysis
4.8. Dual-Luciferase Reporter Assays
4.9. Tumor Implantation and Treatment
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Lozada-Delgado, E.L.; Grafals-Ruiz, N.; Vivas-Mejía, P.E. RNA interference for glioblastoma therapy: Innovation ladder from the bench to clinical trials. Life Sci. 2017, 188, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Shugg, D.; Allen, B.J.; Blizzard, L.; Dwyer, T.; Roder, D. Brain cancer incidence, mortality and case survival: Observations from two australian cancer registries. Int. J. Cancer 1994, 59, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Kleihues, P.; Burger, P.C.; Scheithauer, B.W. The new who classification of brain tumours. Brain Pathol. 1993, 3, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.; Kanu, O.O.; Mehta, A.I.; Di, C.; Lin, N.; Mattox, A.K.; Bigner, D.D. Glioblastoma multiforme: A review of where we have been and where we are going. Expert Opin. Investig. Drugs 2009, 18, 1061–1083. [Google Scholar] [CrossRef] [PubMed]
- Sathornsumetee, S.; Reardon, D.A.; Desjardins, A.; Quinn, J.A.; Vredenburgh, J.J.; Rich, J.N. Molecularly targeted therapy for malignant glioma. Cancer 2007, 110, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Shi, Z.M.; Jiang, C.F.; Liu, X.; Chen, Q.D.; Qian, X.; Li, D.M.; Ge, X.; Wang, X.F.; Liu, L.Z.; et al. Mir-143 acts as a tumor suppressor by targeting n-ras and enhances temozolomide-induced apoptosis in glioma. Oncotarget 2014, 5, 5416–5427. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. Micrornas to nanog, oct4 and sox2 coding regions modulate embryonic stem cell differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Xu, Y.; Xie, X.; Wang, T.; Ko, J.H.; Zhou, T. The role of rna structure at 5’ untranslated region in microrna-mediated gene regulation. RNA 2014, 20, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, T.; Jiang, Y.; Yan, Y.; Qin, R.; Chen, J. Micrornas in human glioblastoma: From bench to beside. Front. Biosci. 2015, 20, 105–118. [Google Scholar]
- Genovese, G.; Ergun, A.; Shukla, S.A.; Campos, B.; Hanna, J.; Ghosh, P.; Quayle, S.N.; Rai, K.; Colla, S.; Ying, H.; et al. Microrna regulatory network inference identifies mir-34a as a novel regulator of tgf-β signaling in glioblastoma. Cancer Discov. 2012, 2, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Díaz, M.; Miranda-Román, M.A.; Soto, D.; Quintero-Aguilo, M.; Ortiz-Zuazaga, H.; Marcos-Martinez, M.J.; Vivas-Mejía, P.E. Microrna-27a distinguishes glioblastoma multiforme from diffuse and anaplastic astrocytomas and has prognostic value. Am. J. Cancer Res. 2015, 5, 201–218. [Google Scholar] [PubMed]
- Iio, A.; Nakagawa, Y.; Hirata, I.; Naoe, T.; Akao, Y. Identification of non-coding rnas embracing microrna-143/145 cluster. Mol. Cancer 2010, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.; Martin, G.S.; Schulz, K.J.; Ronck, M.; Toussaint, L.G. Serial selection for invasiveness increases expression of mir-143/mir-145 in glioblastoma cell lines. BMC Cancer 2012, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Kent, O.A.; McCall, M.N.; Cornish, T.C.; Halushka, M.K. Lessons from mir-143/145: The importance of cell-type localization of mirnas. Nucleic Acids Res. 2014, 42, 7528–7538. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Yadav, T.; Rani, V. Exploring mirna based approaches in cancer diagnostics and therapeutics. Crit. Rev. Oncol. Hematol. 2016, 98, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Niu, X.; Zhang, X.; Tao, J.; Wu, D.; Wang, Z.; Li, P.; Zhang, W.; Wu, H.; Feng, N.; et al. Mir-143 decreases prostate cancer cells proliferation and migration and enhances their sensitivity to docetaxel through suppression of kras. Mol. Cell. Biochem. 2011, 350, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Smyth, P.; Flavin, R.; Cahill, S.; Denning, K.; Aherne, S.; Guenther, S.M.; O’Leary, J.J.; Sheils, O. Comparison of mirna expression patterns using total rna extracted from matched samples of formalin-fixed paraffin-embedded (ffpe) cells and snap frozen cells. BMC Biotechnol. 2007, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Nakajima, G.; Gavin, E.; Morris, C.G.; Kudo, K.; Hayashi, K.; Ju, J. Systematic analysis of microrna expression of rna extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA 2007, 13, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. Mir-145 and mir-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Kondo, S.; Ogata, M.; Kanemoto, S.; Saito, A.; Wanaka, A.; Imaizumi, K. Cleavage of the membrane-bound transcription factor oasis in response to endoplasmic reticulum stress. J. Neurochem. 2006, 96, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Chimienti, F.; Devergnas, S.; Favier, A.; Seve, M. Identification and cloning of a beta-cell-specific zinc transporter, znt-8, localized into insulin secretory granules. Diabetes 2004, 53, 2330–2337. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- von der Mark, H.; Williams, I.; Wendler, O.; Sorokin, L.; von der Mark, K.; Pöschl, E. Alternative splice variants of alpha 7 beta 1 integrin selectively recognize different laminin isoforms. J. Biol. Chem. 2002, 277, 6012–6016. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Yu, Y.P.; Tseng, G.C.; Wu, C.; Chen, K.; Rao, U.N.; Nelson, J.; Michalopoulos, G.K.; Luo, J.H. Analysis of integrin alpha7 mutations in prostate cancer, liver cancer, glioblastoma multiforme, and leiomyosarcoma. J. Natl. Cancer Inst. 2007, 99, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Sethi, M.K.; Buettner, F.F.; Krylov, V.B.; Takeuchi, H.; Nifantiev, N.E.; Haltiwanger, R.S.; Gerardy-Schahn, R.; Bakker, H. Identification of glycosyltransferase 8 family members as xylosyltransferases acting on o-glucosylated notch epidermal growth factor repeats. J. Biol. Chem. 2010, 285, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Yeckel, M.F.; Koleske, A.J. Abl2/arg controls dendritic spine and dendrite arbor stability via distinct cytoskeletal control pathways. J. Neurosci. 2013, 33, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- Procaccia, V.; Nakayama, H.; Shimizu, A.; Klagsbrun, M. Gleevec/imatinib, an abl2 kinase inhibitor, protects tumor and endothelial cells from semaphorin-induced cytoskeleton collapse and loss of cell motility. Biochem. Biophys. Res. Commun. 2014, 448, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Xing, Q.T.; Qu, C.M.; Wang, G. Overexpression of abl2 predicts poor prognosis in hepatocellular carcinomas and is associated with cancer cell migration and invasion. Onco Targets Ther. 2014, 7, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, A.; Rebollo, A. Induction of p53-independent apoptosis by the bh3-only protein itm2bs. FEBS Lett. 2004, 557, 283–287. [Google Scholar] [CrossRef]
- Vidal, R.; Frangione, B.; Rostagno, A.; Mead, S.; Révész, T.; Plant, G.; Ghiso, J. A stop-codon mutation in the bri gene associated with familial british dementia. Nature 1999, 399, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Tamayev, R.; Giliberto, L.; Li, W.; d’Abramo, C.; Arancio, O.; Vidal, R.; D’Adamio, L. Memory deficits due to familial british dementia bri2 mutation are caused by loss of bri2 function rather than amyloidosis. J. Neurosci. 2010, 30, 14915–14924. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, A.; Ayllon, V.; Rebollo, A. Itm2bs regulates apoptosis by inducing loss of mitochondrial membrane potential. Eur. J. Immunol. 2002, 32, 3498–3505. [Google Scholar] [CrossRef]
- Reyes-González, J.M.; Armaiz-Peña, G.N.; Mangala, L.S.; Valiyeva, F.; Ivan, C.; Pradeep, S.; Echevarría-Vargas, I.M.; Rivera-Reyes, A.; Sood, A.K.; Vivas-Mejía, P.E. Targeting c-myc in platinum-resistant ovarian cancer. Mol. Cancer Ther. 2015, 14, 2260–2269. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Gocheva, V.; Bhutkar, A.; Resnick, R.; Jong, R.M.; Miller, K.M.; Bendor, J.; Jacks, T. Stromal expression of mir-143/145 promotes neoangiogenesis in lung cancer development. Cancer Discov. 2016, 6, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.Z.; O’ Connor, S.M.; van Holst Pellekaan, N.G.; Young, G.P.; James, R.J. Reduced accumulation of specific micrornas in colorectal neoplasia. Mol. Cancer Res. 2003, 1, 882–891. [Google Scholar] [PubMed]
- Takagi, T.; Iio, A.; Nakagawa, Y.; Naoe, T.; Tanigawa, N.; Akao, Y. Decreased expression of microrna-143 and -145 in human gastric cancers. Oncology 2009, 77, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Sun, L. Determination of internal controls for quantitative real time rt-pcr analysis of the effect of edwardsiella tarda infection on gene expression in turbot (scophthalmus maximus). Fish Shellfish Immunol. 2011, 30, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, H.; Zhao, T.; Ma, W.; Dong, J.; Zhang, S.; Xin, W.; Yang, S.; Jia, L.; Hao, J. Single nucleotide polymorphism in the microrna-199a binding site of hif1a gene is associated with pancreatic ductal adenocarcinoma risk and worse clinical outcomes. Oncotarget 2016, 7, 13717–13729. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Cui, Z.; Ren, Y.; Xia, L.; Wang, Q.; Zhang, Y.; He, Q.; Zhou, B. Association between polymorphisms in pre-mirna genes and risk of lung cancer in a chinese non-smoking female population. Lung Cancer 2016, 94, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Alves, T.R.; Lima, F.R.; Kahn, S.A.; Lobo, D.; Dubois, L.G.; Soletti, R.; Borges, H.; Neto, V.M. Glioblastoma cells: A heterogeneous and fatal tumor interacting with the parenchyma. Life Sci. 2011, 89, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhang, L. Puma, a potent killer with or without p53. Oncogene 2008, 27, S71–S83. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, Y.; Hua, J.; Yang, X.; Zhang, X.; Duan, M.; Zhu, X.; Huang, W.; Chao, J.; Zhou, R.; et al. Silencing microrna-143 protects the integrity of the blood-brain barrier: Implications for methamphetamine abuse. Sci. Rep. 2016, 6, 35642. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Kanzawa, T.; Miyoshi, T.; Hirohata, S.; Kyo, S.; Iwamaru, A.; Aoki, H.; Kondo, Y.; Kondo, S. Therapeutic efficacy of puma for malignant glioma cells regardless of p53 status. Hum. Gene Ther. 2005, 16, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Merriman, C.; Huang, Q.; Rutter, G.A.; Fu, D. Lipid-tuned zinc transport activity of human znt8 protein correlates with risk for type-2 diabetes. J. Biol. Chem. 2016, 291, 26950–26957. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Fan, J.; Chen, N. A novel regulator of type ii diabetes: Microrna-143. Trends Endocrinol. Metab. 2018, 29, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, L.F.; Zhang, H.W.; Hu, S.; Lu, M.H.; Liang, S.; Li, B.; Li, Y.; Li, D.; Wang, E.D.; et al. A novel mir-155/mir-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012, 31, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Xiao, T.; Fang, Z.; Sun, Y.; Li, F.; Gao, Y.; Feng, Y.; Li, L.; Wang, Y.; Liu, X.; et al. Microrna-143 (mir-143) regulates cancer glycolysis via targeting hexokinase 2 gene. J. Biol. Chem. 2012, 287, 23227–23235. [Google Scholar] [CrossRef] [PubMed]
- Peschiaroli, A.; Giacobbe, A.; Formosa, A.; Markert, E.K.; Bongiorno-Borbone, L.; Levine, A.J.; Candi, E.; D’Alessandro, A.; Zolla, L.; Finazzi Agrò, A.; et al. Mir-143 regulates hexokinase 2 expression in cancer cells. Oncogene 2013, 32, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, L.H.; Jacobsen, A.; Frankel, L.B.; Wen, J.; Krogh, A.; Lund, A.H. Microrna-143 down-regulates hexokinase 2 in colon cancer cells. BMC Cancer 2012, 12, 232. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liu, H.; Liu, Y.; Wu, J.; Wang, C.; Hou, X.; Chen, X.; Yang, G.; Zhao, L.; Che, H.; et al. Mir-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013, 333, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, Y.; Han, B.; Yang, L.; Chen, X.; Huang, R.; Wu, F.; Chao, J.; Liu, P.; Hu, G.; et al. Circular rna dlgap4 ameliorates ischemic stroke outcomes by targeting mir-143 to regulate endothelial-mesenchymal transition associated with blood-brain barrier integrity. J. Neurosci. 2018, 38, 32–50. [Google Scholar] [PubMed]
- Du, C.; Shen, Z.; Zang, R.; Xie, H.; Li, H.; Chen, P.; Hang, B.; Xu, X.; Tang, W.; Xia, Y. Negative feedback circuitry between mir143hg and rbm24 in hirschsprung disease. Biochim. Biophys. Acta 2016, 1862, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Gonzalez, J.M.; Pietri-Vazquez, F.M.; Vivas-Mejia, P.E. A general overview of the nano-sized carriers for cancer treatment. In Nano Based Drug Delivery; Naik, J., Ed.; IAPC Publishing: Zagreb, Croacia, 2015; pp. 317–333. [Google Scholar]
- Echevarría-Vargas, I.M.; Valiyeva, F.; Vivas-Mejía, P.E. Upregulation of mir-21 in cisplatin resistant ovarian cancer via jnk-1/c-jun pathway. PLoS ONE 2014, 9, e97094. [Google Scholar] [CrossRef] [PubMed]
- Vargas, I.M.; Vivas-Mejía, P.E. Assessment of mrna splice variants by qrt-pcr. Methods Mol. Biol. 2013, 1049, 171–186. [Google Scholar] [PubMed]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Full Name | Gene Symbol | Fold Change miR-143-Inh vs. NC-Inh in T98G Cells | Biological Role |
|---|---|---|---|
| Solute carrier family 30 (zinc transporter), member 8 | SLC30A8 | 1.98 | Role in glucose homeostasis; diabetes mellitus [25,26]. |
| Integrin, alpha 7 | ITGA7 | 1.51 | Receptor for the basement membrane protein laminin-1 [27]; Tumor suppressor gene [28]. |
| Glucoside xylosyltransferase 1 | GXYLT1 | 1.69 | Notch xylosyltransferase [29]. |
| ABL proto-oncogene 2, non-receptor tyrosine kinase | ABL2 | 1.53 | Oncogene related to cell migration and invasion in various cancers; role in cytoskeletal rearrangement, regulator of neuronal structural stability [30,31,32]. |
| Integral membrane protein 2B | ITM2B | 1.32 | Role in triggering apoptosis P53 independent; tumor suppressor [33]. Transmembrane protein involved in negative regulation of amyloid processing. Related to dementia [34,35,36]. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozada-Delgado, E.L.; Grafals-Ruiz, N.; Miranda-Román, M.A.; Santana-Rivera, Y.; Valiyeva, F.; Rivera-Díaz, M.; Marcos-Martínez, M.J.; Vivas-Mejía, P.E. Targeting MicroRNA-143 Leads to Inhibition of Glioblastoma Tumor Progression. Cancers 2018, 10, 382. https://doi.org/10.3390/cancers10100382
Lozada-Delgado EL, Grafals-Ruiz N, Miranda-Román MA, Santana-Rivera Y, Valiyeva F, Rivera-Díaz M, Marcos-Martínez MJ, Vivas-Mejía PE. Targeting MicroRNA-143 Leads to Inhibition of Glioblastoma Tumor Progression. Cancers. 2018; 10(10):382. https://doi.org/10.3390/cancers10100382
Chicago/Turabian StyleLozada-Delgado, Eunice L., Nilmary Grafals-Ruiz, Miguel A. Miranda-Román, Yasmarie Santana-Rivera, Fatma Valiyeva, Mónica Rivera-Díaz, María J. Marcos-Martínez, and Pablo E. Vivas-Mejía. 2018. "Targeting MicroRNA-143 Leads to Inhibition of Glioblastoma Tumor Progression" Cancers 10, no. 10: 382. https://doi.org/10.3390/cancers10100382