Sensitization of Cancer Cells to Radiation and Topoisomerase I Inhibitor Camptothecin Using Inhibitors of PARP and Other Signaling Molecules

{kind=link}
{kind=link}
Abstract
:1. Introduction
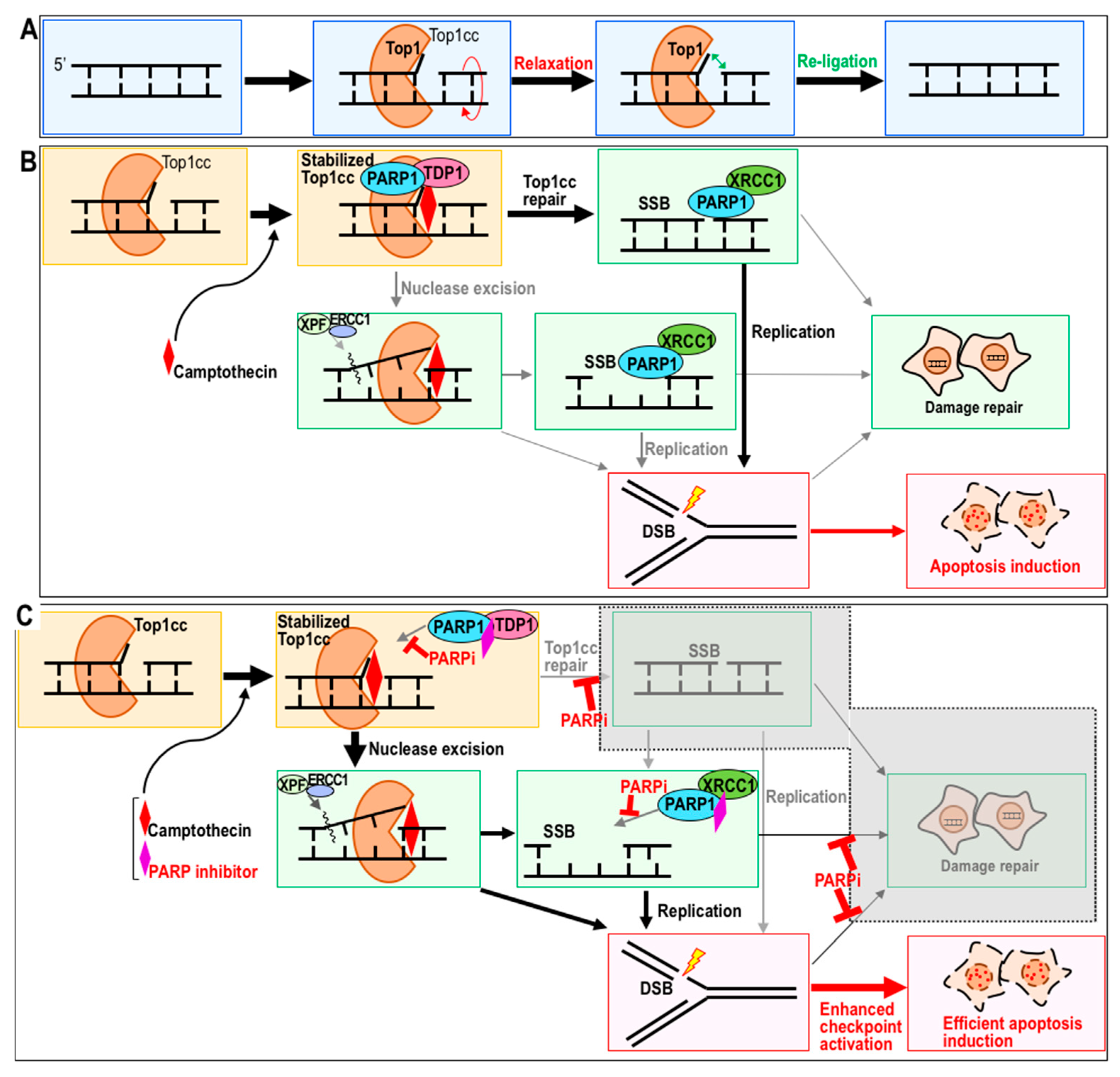
2. PARP Inhibitor as a Potential Sensitizer to Top1 Inhibitor
2.1. Top1 Inhibitor Treatment in the Presence of PARP Inhibitor
2.2. Sensitization to CPT by PARP Inhibitor
2.3. Potential Combination Therapy with CPT and a PARP Inhibitor as a Sensitizer
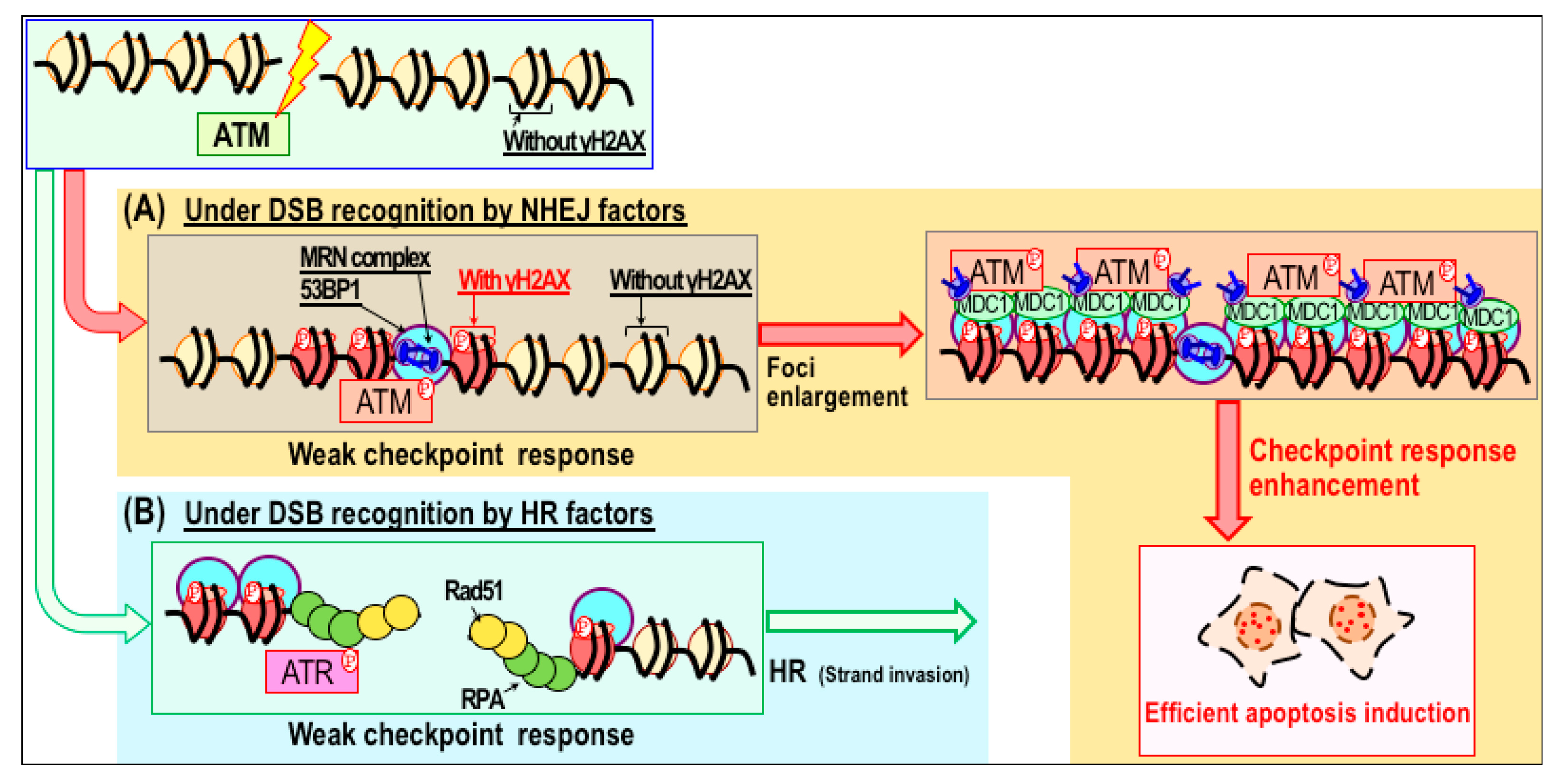
3. Radiation Therapy and Its Sensitizers
3.1. Radiosensitizers and Their Clinical Use
3.2. Sensitization to Radiation Therapy through Modulation of Repair Pathways
3.3. Radiotherapy in Conjunction with Molecularly Targeted Agents and Immune Checkpoint Inhibitors
3.4. Radiosensitivity by Autophagy Regulatory Drugs
3.5. Proton Beam and Carbon-Ion Beam Therapy
4. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Von Sonntag, C. Free DNA Damage Its Repair a Chemistry Perspect; Springer: Berlin, Germany, 2006; pp. 7–46. [Google Scholar]
- Adhikary, A.; Becker, D.; Sevilla, M.D. Electron Spin Resonance of Radicals in Irradiated DNA; Springer: Berlin, Germany, 2014; pp. 299–352. [Google Scholar]
- Yu, Q.; Rose, J.H.L.; Zhang, H.; Pommier, Y. Antisense inhibition of Chk2/hCds1 expression attenuates DNA damage-induced S and G2 checkpoints and enhances apoptotic activity in HEK-293 cells. FEBS Lett. 2001, 505, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Hiu, W.C.; Jin, D.Y.; Ling, M.T.; Yong, C.W.; Wang, Q.; Sai, W.T.; Wang, X. Mitotic arrest deficient 2 expression induces chemosensitization to a DNA-damaging agent, cisplatin, in nasopharyngeal carcinoma cells. Cancer Res. 2005, 65, 1450–1458. [Google Scholar]
- Attardi, L.D.; de Vries, A.; Jacks, T. Activation of the p53-dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene 2004, 23, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, J.; DeStephanis, D.; Patel, Y.; Gowda, V.; Yan, S. Study of the DNA damage checkpoint using Xenopus egg extracts. J. Vis. Exp. 2012, 69, e4449. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, M.; Kalinowska, M.; Skierski, J.; Lewandowski, W. A review of selected anti-tumour therapeutic agents and reasons for multidrug resistance occurrence. J. Pharm. Pharmacol. 2004, 56, 1067–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, e28. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, X.; Le Tourneau, C.; Verweij, J.; Siu, L.L.; Seymour, L.; Postel-Vinay, S.; Collette, L.; Rizzo, E.; Ivy, P.; Olmos, D.; et al. Defining dose-limiting toxicity for phase 1 trials of molecularly targeted agents: Results of a DLT-TARGETT international survey. Eur. J. Cancer 2014, 50, 2050–2056. [Google Scholar] [CrossRef] [PubMed]
- Bartz, S.R.; Zhang, Z.; Burchard, J.; Imakura, M.; Martin, M.; Palmieri, A.; Needham, R.; Guo, J.; Gordon, M.; Chung, N.; et al. Small Interfering RNA Screens Reveal Enhanced Cisplatin Cytotoxicity in Tumor Cells Having both BRCA Network and TP53 Disruptions. Mol. Cell. Biol. 2006, 26, 9377–9386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van Der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engert, F.; Schneider, C.; Weiβ, L.M.; Probst, M.; Fulda, S. PARP Inhibitors Sensitize Ewing Sarcoma Cells to Temozolomide-Induced Apoptosis via the Mitochondrial Pathway. Mol. Cancer Ther. 2015, 14, 2818–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, S.J.; Travers, J.; Pshenichnaya, I.; Kogera, F.A.; Barthorpe, S.; Mironenko, T.; Richardson, L.; Benes, C.H.; Stratton, M.R.; McDermott, U.; et al. Combinations of PARP Inhibitors with Temozolomide Drive PARP1 Trapping and Apoptosis in Ewing’s Sarcoma. PLoS ONE 2015, 10, e0140988. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, A.J. The potential role and application of PARP inhibitors in cancer treatment. Br. Med. Bull. 2009, 89, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.M.; Szopa, J.; Han, F.S.; Cheng, Y.C.; Richter, A.; Scheer, U. Association of DNA Topoisomerase I and RNA Polymerase I: A Possible Role for Topoisomerase I in Ribosomal Gene Transcription; Springer: Berlin, Germany, 1988; pp. 411–416. [Google Scholar]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, M.E.; Warn, M.C. Camptothecin and Taxol: Discovery to Clinic—Thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1995, 55, 753–760. [Google Scholar] [PubMed]
- Venditto, V.J.; Simanek, E.E. Cancer therapies utilizing the camptothecins: A review of the in vivo literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Das, B.B.; Huang, S.N.; Murai, J.; Rehman, I.; Amé, J.-C.C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, K.; Takebayashi, S.-I.; Taguchi, H.; Takeda, S.; Okumura, K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008, 183, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Barcelo, J.M.; Rao, V.A.; Sordet, O.; Jobson, A.G.; Thibaut, L.; Miao, Z.-H.; Seiler, J.A.; Zhang, H.; Marchand, C.; et al. Repair of topoisomerase I—Mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 179–229. [Google Scholar] [PubMed]
- Pommier, Y. Drugging topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.A.; Anarbaev, R.O.; Sukhanova, M.; Vasil’eva, I.A.; Rechkunova, N.I.; Lavrik, O.I. Poly(ADP-ribose)polymerase 1 stimulates the AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1. Biosci. Rep. 2015, 35, e00230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Regairaz, M.; Seiler, J.; Agama, K.K.; Doroshow, J.H.; Pommier, Y. Poly (ADP-ribose) polymerase and XPF–ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011, 39, 3607–3620. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Redon, C.; Rao, V.A.; Seiler, J.A.; Sordet, O.; Takemura, H.; Antony, S.; Meng, L.; Liao, Z.; Kohlhagen, G.; et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat. Res. Mol. Mech. Mutagen. 2003, 532, 173–203. [Google Scholar] [CrossRef] [Green Version]
- Konecny, G.E.; Kristeleit, R.S. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: Current practice and future directions. Br. J. Cancer 2016, 115, 1157–1173. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; Flatten, K.S.; Schneider, P.A.; Dai, N.T.; McDonald, J.S.; Poirier, G.G.; Kaufmann, S.H. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. J. Biol. Chem. 2012, 287, 4198–4210. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Rehman, I.; Ghosh, A.; Sengupta, S.; Majumdar, P.; Jana, B.; Das, B.B. Poly(ADP-ribose) polymers regulate DNA topoisomerase i (Top1) nuclear dynamics and camptothecin sensitivity in living cells. Nucleic Acids Res. 2016, 44, 8363–8375. [Google Scholar] [CrossRef] [PubMed]
- Ström, C.E.; Johansson, F.; Uhlén, M.; Szigyarto, C.A.K.; Erixon, K.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef] [PubMed]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsumi, Y.; Inase, A.; Osawa, T.; Sugihara, E.; Sakasai, R.; Fujimori, H.; Teraoka, H.; Saya, H.; Kanno, M.; Tashiro, F.; et al. The Arf/p53 protein module, which induces apoptosis, down-regulates histone H2AX to allow normal cells to survive in the presence of anti-cancer drugs. J. Biol. Chem. 2013, 288, 13269–13277. [Google Scholar] [CrossRef] [PubMed]
- Gartner, A.; Milstein, S.; Ahmed, S.; Hodgkin, J.; Hengartner, M.O. A Conserved Checkpoint Pathway Mediates DNA Damage–Induced Apoptosis and Cell Cycle Arrest in C. elegans. Mol. Cell 2000, 5, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, e12716. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Pires, I.M.; Bencokova, Z.; Coackley, C.; Luoto, K.R.; Bhogal, N.; Lakshman, M.; Gottipati, P.; Oliver, F.J.; Helleday, T.; et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010, 70, 8045–8054. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Do roshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Hirai, T.; Shirai, H.; Fujimori, H.; Okayasu, R.; Sasai, K.; Masutani, M. Radiosensitization effect of poly(ADP-ribose) polymerase inhibition in cells exposed to low and high liner energy transfer radiation. Cancer Sci. 2012, 103, 1045–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samol, J.; Ranson, M.; Scott, E.; Macpherson, E.; Carmichael, J.; Thomas, A.; Cassidy, J. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: A phase I study. Investig. New Drugs 2012, 30, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.; O’Reilly, S. Clinical Guidelines for Managing Topotecan-Related Hematologic Toxicity. Oncologist 1998, 3, 4–10. [Google Scholar] [PubMed]
- Chen, Y.; Pandya, K.; Keng, P.C.; Johnstone, D.; Li, J.; Lee, Y.-J.; Smudzin, T.; Okunieff, P. Phase I/II clinical study of pulsed paclitaxel radiosensitization for thoracic malignancy: A therapeutic approach on the basis of preclinical research of human cancer cell lines. Clin. Cancer Res. 2003, 9, 969–975. [Google Scholar] [PubMed]
- Chinnaiyan, P.; Huang, S.; Vallabhaneni, G.; Armstrong, E.; Varambally, S.; Tomlins, S.A.; Chinnaiyan, A.M.; Harari, P.M. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva). Cancer Res. 2005, 65, 3328–3335. [Google Scholar] [CrossRef] [PubMed]
- van Nifterik, K.A.; van den Berg, J.; Stalpers, L.J.A.; Lafleur, M.V.M.; Leenstra, S.; Slotman, B.J.; Hulsebos, T.J.M.; Sminia, P. Differential Radiosensitizing Potential of Temozolomide in MGMT Promoter Methylated Glioblastoma Multiforme Cell Lines. Int. J. Radiat. Oncol. 2007, 69, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Bobola, M.S.; Kolstoe, D.D.; Blank, A.; Silber, J.R. Minimally cytotoxic doses of temozolomide produce radiosensitization in human glioblastoma cells regardless of MGMT expression. Mol. Cancer Ther. 2010, 9, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, G.; Joensuu, T.; Nokisalmi, P.; Reddy, C.; Isola, J.; Ruutu, M.; Kouri, M.; Kupelian, P.A.; Collan, J.; Pesonen, S.; et al. A Phase I/II Trial of Gefitinib Given Concurrently with Radiotherapy in Patients with Nonmetastatic Prostate Cancer. Int. J. Radiat. Oncol. 2010, 78, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Choy, H.; Safran, H.; Akerley, W.; Graziano, S.L.; Bogart, J.A.; Cole, B.F. Phase II trial of weekly paclitaxel and concurrent radiation therapy for locally advanced non-small cell lung cancer. Clin. Cancer Res. 1998, 4, 1931–1936. [Google Scholar] [PubMed]
- Lau, D.; Ryu, J.; Gandara, D.; Morgan, R.; Doroshow, J.; Wilder, R.; Leigh, B. Concurrent twice-weekly paclitaxel and thoracic irradiation for stage III non-small cell lung cancer. Semin. Radiat. Oncol. 1999, 9, 117–120. [Google Scholar] [PubMed]
- Keys, H.M.; Bundy, B.N.; Stehman, F.B.; Muderspach, L.I.; Chafe, W.E.; Suggs, C.L.; Walker, J.L.; Gersell, D. Cisplatin, Radiation, and Adjuvant Hysterectomy Compared with Radiation and Adjuvant Hysterectomy for Bulky Stage IB Cervical Carcinoma. N. Engl. J. Med. 1999, 340, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent Cisplatin-Based Radiotherapy and Chemotherapy for Locally Advanced Cervical Cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Boeckman, H.J. Cisplatin Sensitizes Cancer Cells to Ionizing Radiation via Inhibition of Nonhomologous End Joining. Mol. Cancer Res. 2005, 3, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Ren, Y.; Zhang, T.; Wang, Z.; Ling, C.C.; Li, G.C.; He, F.; Wang, C.; Wen, B. Inactivation of DNA-PK by knockdown DNA-PKcs or NU7441 impairs non-homologous end-joining of radiation-induced double strand break repair. Oncol. Rep. 2018, 39, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, C.V.M.; de Haan, R.; Hageman, F.; Oostendorp, T.P.D.; Carli, A.L.E.; O’Connor, M.J.; Jonkers, J.; Verheij, M.; van den Brekel, M.W.; Vens, C. Extent of radiosensitization by the PARP inhibitor olaparib depends on its dose, the radiation dose and the integrity of the homologous recombination pathway of tumor cells. Radiother. Oncol. 2015, 116, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, M.; Sharma, K.; Saleh, T.; Povirk, L.F.; Hendrickson, E.A.; Gewirtz, D.A. Radiosensitization by PARP Inhibition in DNA Repair Proficient and Deficient Tumor Cells: Proliferative Recovery in Senescent Cells. Radiat. Res. 2016, 185, 229–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Quesada, R.; Muñoz-Gámez, J.; Martín-Oliva, D.; Peralta, A.; Valenzuela, M.T.; Matínez-Romero, R.; Quiles-Pérez, R.; Murcia, J.; de Murcia, G.; de Almodóvar, M.R.; et al. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol. Biol. 2007, 8, e29. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.; Hatahet, Z.; Wallace, S.S. In vitro repair of synthetic ionizing radiation-induced multiply damaged DNA sites. J. Mol. Biol. 1999, 290, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisz, J.A.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C.M. Effects of ionizing radiation on biological molecules—Mechanisms of damage and emerging methods of detection. Antioxid. Redox Signal. 2014, 21, 260–292. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological Consequences of Radiation-induced DNA Damage: Relevance to Radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Yang, E.S. The intersection between DNA damage response and cell death pathways. Exp. Oncol. 2012, 34, 243–254. [Google Scholar] [PubMed]
- Atsumi, Y.; Minakawa, Y.; Ono, M.; Dobashi, S.; Shinohe, K.; Shinohara, A.; Takeda, S.; Takagi, M.; Takamatsu, N.; Nakagama, H.; et al. ATM and SIRT6/SNF2H Mediate Transient H2AX Stabilization When DSBs Form by Blocking HUWE1 to Allow Efficient γH2AX Foci Formation. Cell Rep. 2015, 13, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, Y.; Atsumi, Y.; Shinohara, A.; Murakami, Y.; Yoshioka, K. Gamma-irradiated quiescent cells repair directly induced double-strand breaks but accumulate persistent double-strand breaks during subsequent DNA replication. Genes Cells 2016, 21, 789–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Liang, K.; Lu, Y.; Fan, Z. The anti-EGFR antibody cetuximab sensitizes human head and neck squamous cell carcinoma cells to radiation in part through inhibiting radiation-induced upregulation of HIF-1alpha. Cancer Let. 2012, 322, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, I.; Hagekyriakou, J.; Sharp, L.L.; Galli, M.; West, A.; McLaughlin, N.M.; Duret, H.; Yagita, H.; Johnstone, R.W.; Smyth, M.J.; et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res. 2012, 72, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining Radiotherapy and Cancer Immunotherapy: A Paradigm Shift. JNCI J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Sharma, R.; Chin, R.; Tu, T.; Weichselbaum, R.R.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, S.; Comincini, S. Autophagy and ionizing radiation in tumors: The “survive or not survive” dilemma. J. Cell. Physiol. 2013, 228, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Daido, S.; Yamamoto, A.; Fujiwara, K.; Sawaya, R.; Kondo, S.; Kondo, Y. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005, 65, 4368–4375. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ma, S.; Liu, M.; Hou, Y.; Liang, B.; Su, X.; Liu, X. Synergistic killing of lung cancer cells by cisplatin and radiation via autophagy and apoptosis. Oncol. Lett. 2014, 7, 1903–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.; Wang, Y.; Pang, X.; Zhang, B. Inhibition of autophagy induced by TSA sensitizes colon cancer cell to radiation. Tumor Biol. 2014, 35, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Jiang, F.; Yang, C.; Yan, Q.; Guo, W.; Huang, Q.; Zhang, L.; Jiang, G. Role of autophagy in regulating the radiosensitivity of tumor cells. J. Cancer Res. Clin. Oncol. 2017, 143, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Ishikawa, H.; Okumura, T. Proton beam therapy in Japan: Current and future status. Jpn. J. Clin. Oncol. 2016, 46, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Alan Mitteer, R.; Wang, Y.; Shah, J.; Gordon, S.; Fager, M.; Butter, P.-P.; Jun Kim, H.; Guardiola-Salmeron, C.; Carabe-Fernandez, A.; Fan, Y. Proton beam radiation induces DNA damage and cell apoptosis in glioma stem cells through reactive oxygen species. Sci. Rep. 2015, 5, e13961. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; David, G.H.; Piero, D.; Charles, F.B.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Sai, S.; Oonishi, K.; Yamada, S.; Kamada, T.; Okayasu, R. Effects of carbon ion beam on putative colon cancer stem cells and its correlation with radiocurability. Radiother. Oncol. 2011, 99, e393. [Google Scholar] [CrossRef]
- Mohamad, O.; Sishc, B.J.; Saha, J.; Pompos, A.; Rahimi, A.; Story, M.D.; Davis, A.J.; Kim, D.W.N. Carbon ion radiotherapy: A review of clinical experiences and preclinical research, with an emphasis on DNA damage/repair. Cancers 2017, 9, e66. [Google Scholar] [CrossRef] [PubMed]
- Ibañez, I.L.; Bracalente, C.; Molinari, B.L.; Palmieri, M.A.; Policastro, L.; Kreiner, A.J.; Burlón, A.A.; Valda, A.; Navalesi, D.; Davidson, J.; et al. Induction and Rejoining of DNA Double Strand Breaks Assessed by H2AX Phosphorylation in Melanoma Cells Irradiated with Proton and Lithium Beams. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Oonishi, K.; Cui, X.; Hirakawa, H.; Fujimori, A.; Kamijo, T.; Yamada, S.; Yokosuka, O.; Kamada, T. Different effects of carbon ion beams and X-rays on clonogenic survival and DNA repair in human pancreatic cancer stem-like cells. Radiother. Oncol. 2012, 105, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The Major DNA Repair Pathway after Both Proton and Carbon-Ion Radiation is NHEJ, but the HR Pathway is More Relevant in Carbon Ions. Radiat. Res. 2015, 183, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, N.; Noda, S.; Takahashi, A.; Yoshida, Y.; Oike, T.; Murata, K.; Musha, A.; Suzuki, Y.; Ohno, T.; Takahashi, T.; et al. Radiosensitizing effect of carboplatin and paclitaxel to carbon-ion beam irradiation in the non-small-cell lung cancer cell line H460. J. Radiat. Res. 2015, 56, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.K.; Matsumoto, Y.; Furusawa, Y.; Kamada, T. PU-H71, a novel Hsp90 inhibitor, as a potential cancer-specific sensitizer to carbon-ion beam therapy. J. Radiat. Res. 2016, 57, 572–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, P.; Hirayama, R.; Niu, Y.; Liu, X.; Chen, W.; Jin, X.; Zhang, P.; Ye, F.; Zhao, T.; et al. Genistein sensitizes glioblastoma cells to carbon ions via inhibiting DNA-PKcs phosphorylation and subsequently repressing NHEJ and delaying HR repair pathways. Radiother. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gerelchuluun, A.; Maeda, J.; Manabe, E.; Brents, C.A.; Sakae, T.; Fujimori, A.; Chen, D.J.; Tsuboi, K.; Kato, T.A. Histone Deacetylase Inhibitor Induced Radiation Sensitization Effects on Human Cancer Cells after Photon and Hadron Radiation Exposure. Int. J. Mol. Sci. 2018, 19, e496. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuno, Y.; Hyodo, M.; Fujimori, H.; Shimizu, A.; Yoshioka, K.-i. Sensitization of Cancer Cells to Radiation and Topoisomerase I Inhibitor Camptothecin Using Inhibitors of PARP and Other Signaling Molecules. Cancers 2018, 10, 364. https://doi.org/10.3390/cancers10100364
Matsuno Y, Hyodo M, Fujimori H, Shimizu A, Yoshioka K-i. Sensitization of Cancer Cells to Radiation and Topoisomerase I Inhibitor Camptothecin Using Inhibitors of PARP and Other Signaling Molecules. Cancers. 2018; 10(10):364. https://doi.org/10.3390/cancers10100364
Chicago/Turabian StyleMatsuno, Yusuke, Mai Hyodo, Haruka Fujimori, Atsuhiro Shimizu, and Ken-ichi Yoshioka. 2018. "Sensitization of Cancer Cells to Radiation and Topoisomerase I Inhibitor Camptothecin Using Inhibitors of PARP and Other Signaling Molecules" Cancers 10, no. 10: 364. https://doi.org/10.3390/cancers10100364