HDAC1 and HDAC2 Double Knockout Triggers Cell Apoptosis in Advanced Thyroid Cancer

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
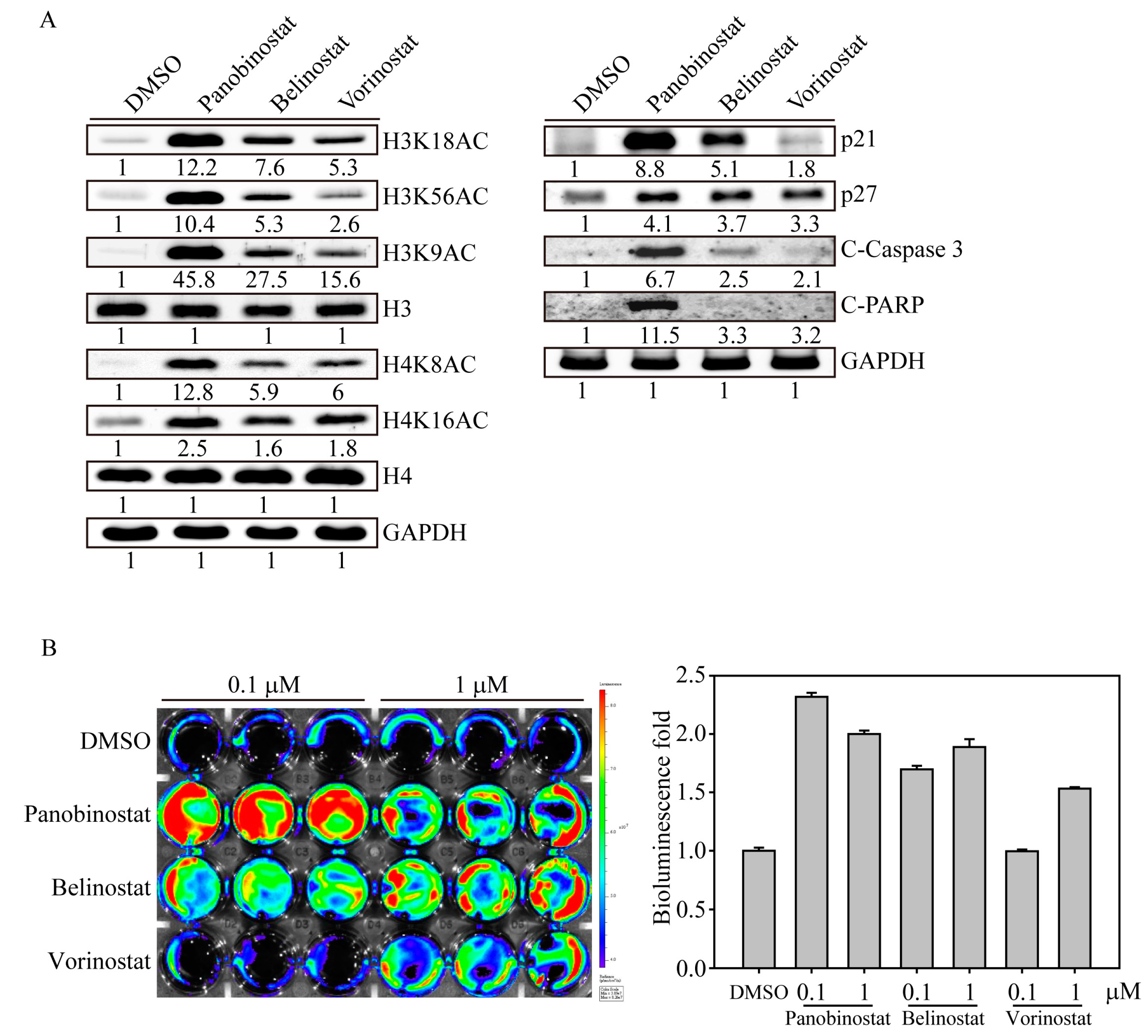
2.1. FDA-Approved HDACi Induced SW579 Cell Apoptosis
2.2. HDACi Induced Histone Acetylation and Apoptosis-Related Protein Expression
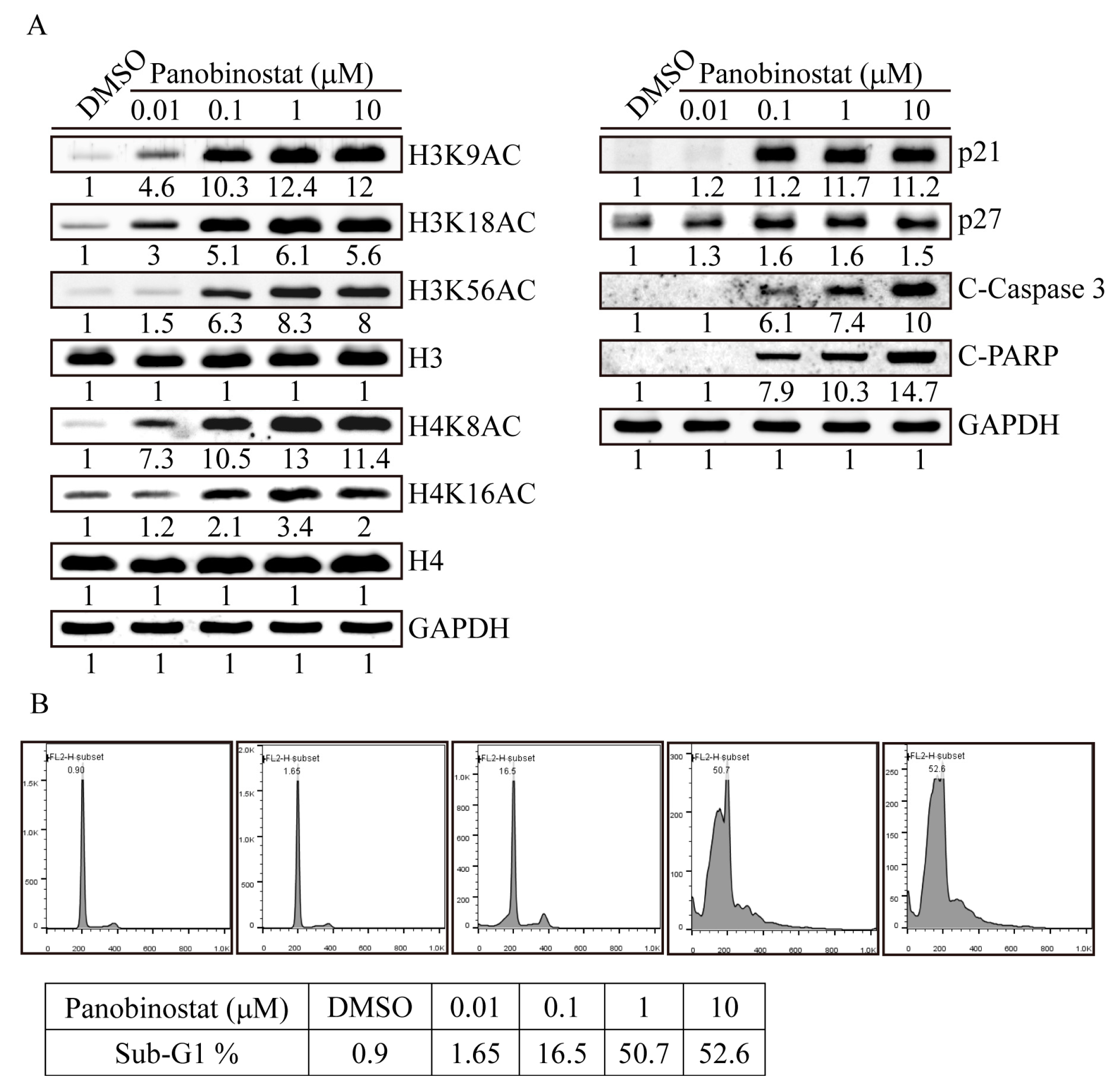
2.3. Panobinostat Dose-Dependently Induced Histone Acetylation and Apoptosis-Related Protein Expression in SW576 Cells
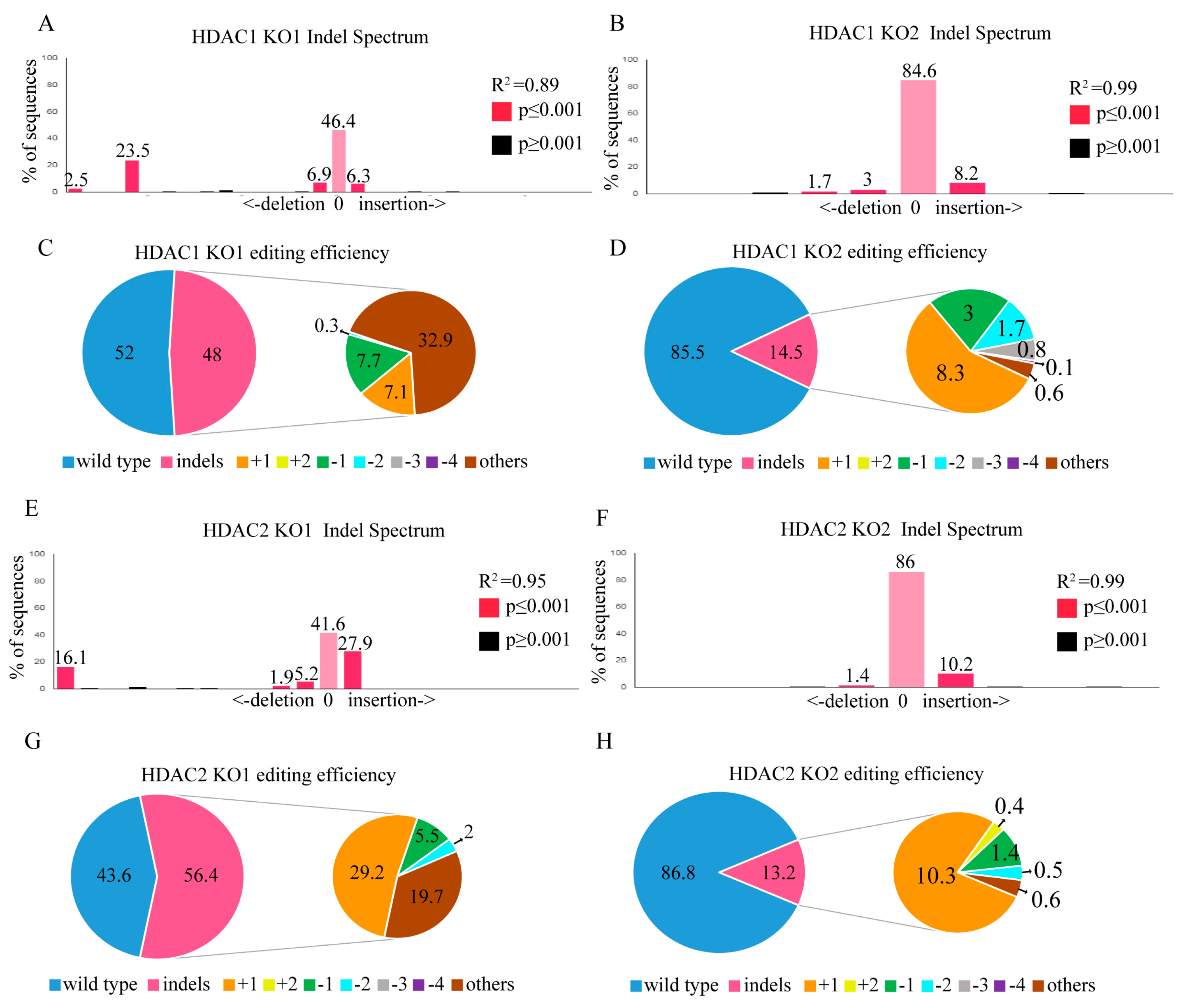
2.4. HDAC1 and HDAC2 Gene Editing through the CRISPR/CAS9 System
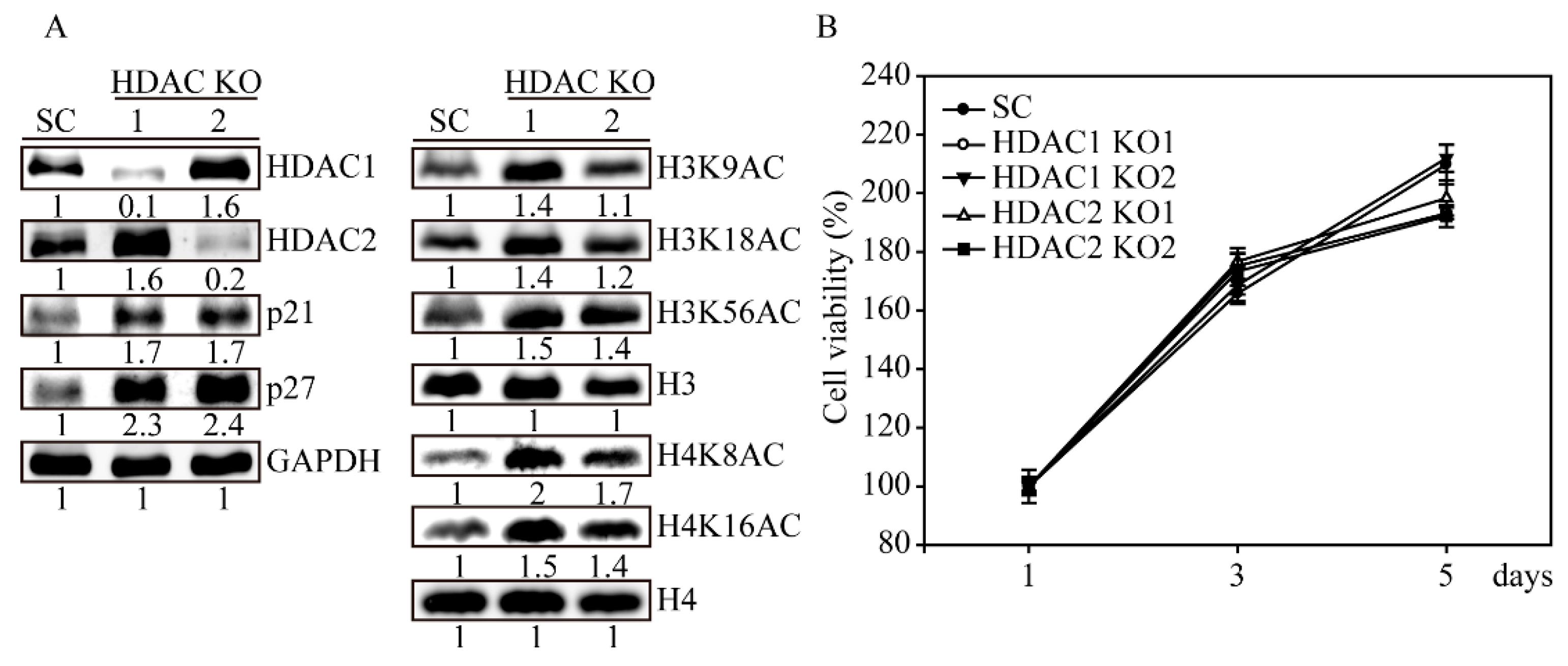
2.5. HDAC1 and HDAC2 Knockout Activates H3 and H4 Histone Acetylations
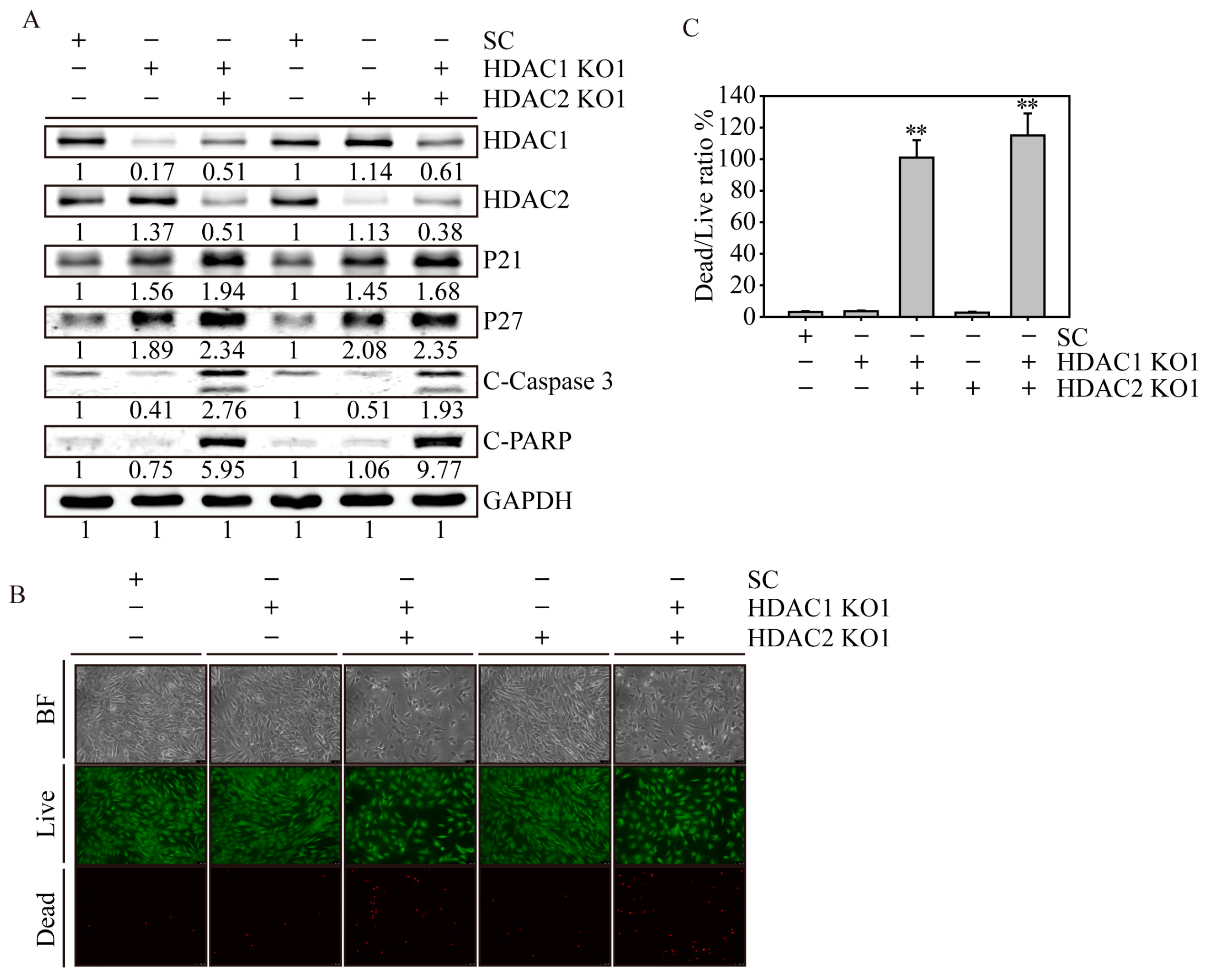
2.6. HDAC1 and HDAC2 Double Knockout Induced Cell Apoptosis in SW579 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. MTT Cell Viability Assay
4.3. Live/Dead Cell Assay
4.4. Cellular Bioluminescence (IVIS) Assay
4.5. Flow Cytometry Analysis
4.6. Protein Extraction, Western Blotting and Antibodies
4.7. Lentivirus Production and Cell Transduction
4.8. Sequencing of Single Guide RNA (sgRNA) Target Sites
4.9. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| STC | Squamous thyroid carcinoma |
| HDACi | Histone deacetylase inhibitor |
| NIADS | Noninvasive apoptosis detection sensor |
| KO | Knockout |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeat |
| IVIS | in vivo imaging system |
References
- Koyama, S.; Fujiwara, K.; Nosaka, K.; Fukuhara, T.; Morisaki, T.; Miyake, N.; Kitano, H.; Takeuchi, H. Immunohistochemical Features of Primary Pure Squamous Cell Carcinoma in the Thyroid: An Autopsy Case. Case Rep. Oncol. 2018, 11, 418–424. [Google Scholar] [CrossRef]
- Keutgen, X.M.; Sadowski, S.M.; Kebebew, E. Management of anaplastic thyroid cancer. Gland Surg. 2015, 4, 44–51. [Google Scholar] [PubMed]
- Struller, F.; Senne, M.; Falch, C.; Kirschniak, A.; Konigsrainer, A.; Muller, S. Primary squamous cell carcinoma of the thyroid: Case report and systematic review of the literature. Int. J. Surg. Case Rep. 2017, 37, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Kebebew, E. Anaplastic thyroid cancer: Rare, fatal and neglected. Surgery 2012, 152, 1088–1089. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, G.; Liu, X.; Li, S. The effects of hyperoside on apoptosis and the expression of Fas/FasL and survivin in SW579 human thyroid squamous cell carcinoma cell line. Oncol. Lett. 2017, 14, 2310–2314. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.; Tulla, K.; Maker, A.V.; Burman, K.D.; Prabhakar, B.S. Therapeutic advances in anaplastic thyroid cancer: A current perspective. Mol. Cancer 2018, 17, 154. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.M. Researches of Epigenetic Epidemiology for Infections and Radiation as Carcinogen. J. Prev. Med. Public Health 2018, 51, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Chmielik, E.; Rusinek, D.; Oczko-Wojciechowska, M.; Jarzab, M.; Krajewska, J.; Czarniecka, A.; Jarzab, B. Heterogeneity of Thyroid Cancer. Pathobiology 2018, 85, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Khatami, F.; Tavangar, S.M. Genetic and Epigenetic of Medullary Thyroid Cancer. Iran Biomed. J. 2018, 22, 142–150. [Google Scholar]
- Sasanakietkul, T.; Murtha, T.D.; Javid, M.; Korah, R.; Carling, T. Epigenetic modifications in poorly differentiated and anaplastic thyroid cancer. Mol. Cell Endocrinol. 2018, 469, 23–37. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, D.; Murugan, A.K.; Liu, Z.; Xing, M. Histone deacetylation of NIS promoter underlies BRAF V600E-promoted NIS silencing in thyroid cancer. Endocr. Relat. Cancer 2014, 21, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, M.; Fortunati, N.; Germano, A.; Asioli, S.; Marano, F.; Palestini, N.; Frairia, R.; Boccuzzi, G.; Catalano, M.G. Histone deacetylase inhibition affects sodium iodide symporter expression and induces 131I cytotoxicity in anaplastic thyroid cancer cells. Thyroid 2013, 23, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.G.; Poli, R.; Pugliese, M.; Fortunati, N.; Boccuzzi, G. Emerging molecular therapies of advanced thyroid cancer. Mol. Aspects Med. 2010, 31, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.W.; Huang, C.Y.; Tam, K.W.; Lin, C.Y.; Huang, L.C.; Lin, C.L.; Hsieh, W.S.; Chi, W.M.; Chang, Y.J.; Wei, P.L.; et al. The Application of Non-Invasive Apoptosis Detection Sensor (NIADS) on Histone Deacetylation Inhibitor (HDACi)-Induced Breast Cancer Cell Death. Int. J. Mol. Sci. 2018, 19, 452. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Su, L.; Xiao, H. Review of Factors Related to the Thyroid Cancer Epidemic. Int. J. Endocrinol. 2017, 2017, 5308635. [Google Scholar] [CrossRef] [PubMed]
- Zarkesh, M.; Zadeh-Vakili, A.; Azizi, F.; Foroughi, F.; Akhavan, M.M.; Hedayati, M. Altered Epigenetic Mechanisms in Thyroid Cancer Subtypes. Mol. Diagn. Ther. 2018, 22, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Stephen, J.K.; Chitale, D.; Narra, V.; Chen, K.M.; Sawhney, R.; Worsham, M.J. DNA methylation in thyroid tumorigenesis. Cancers 2011, 3, 1732–1743. [Google Scholar] [CrossRef]
- Zhu, X.; Cheng, S.Y. Epigenetic Modifications: Novel Therapeutic Approach for Thyroid Cancer. Endocrinol. Metab. 2017, 32, 326–331. [Google Scholar] [CrossRef]
- Catalano, M.G.; Fortunati, N.; Boccuzzi, G. Epigenetics modifications and therapeutic prospects in human thyroid cancer. Front. Endocrinol. 2012, 3, 40. [Google Scholar] [CrossRef]
- Mancikova, V.; Buj, R.; Castelblanco, E.; Inglada-Perez, L.; Diez, A.; de Cubas, A.A.; Curras-Freixes, M.; Maravall, F.X.; Mauricio, D.; Matias-Guiu, X.; et al. DNA methylation profiling of well-differentiated thyroid cancer uncovers markers of recurrence free survival. Int. J. Cancer 2014, 135, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Yee, A.J.; Raje, N.S. Panobinostat and Multiple Myeloma in 2018. Oncologist 2018, 23, 516–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, M.G.; Pugliese, M.; Gargantini, E.; Grange, C.; Bussolati, B.; Asioli, S.; Bosco, O.; Poli, R.; Compagnone, A.; Bandino, A.; et al. Cytotoxic activity of the histone deacetylase inhibitor panobinostat (LBH589) in anaplastic thyroid cancer in vitro and in vivo. Int. J. Cancer 2012, 130, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Gelles, J.D.; Chipuk, J.E. Robust high-throughput kinetic analysis of apoptosis with real-time high-content live-cell imaging. Cell Death Dis. 2016, 7, e2493. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; Zheng, Y.; Tyner, J.W.; Chng, W.J.; Chien, W.W.; Gery, S.; Leong, G.; Braunstein, G.D.; Koeffler, H.P. Belinostat and panobinostat (HDACI): In vitro and in vivo studies in thyroid cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 1507–1514. [Google Scholar] [CrossRef]
- Inoue, S.; Mai, A.; Dyer, M.J.; Cohen, G.M. Inhibition of histone deacetylase class I but not class II is critical for the sensitization of leukemic cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2006, 66, 6785–6792. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Cubizolles, F.; Zhang, Y.; Reichert, N.; Kohler, H.; Seiser, C.; Matthias, P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010, 24, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Wilting, R.H.; Yanover, E.; Heideman, M.R.; Jacobs, H.; Horner, J.; van der Torre, J.; DePinho, R.A.; Dannenberg, J.H. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010, 29, 2586–2597. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Huang, C.S.; Chen, C.S.; Tu, S.H.; Wang, Y.J.; Chang, Y.J.; Tam, K.W.; Wei, P.L.; Cheng, T.C.; Chu, J.S.; et al. Overexpression and activation of the alpha9-nicotinic receptor during tumorigenesis in human breast epithelial cells. J. Natl. Cancer Inst. 2010, 102, 1322–1335. [Google Scholar] [CrossRef]
- Tu, S.H.; Hsieh, Y.C.; Huang, L.C.; Lin, C.Y.; Hsu, K.W.; Hsieh, W.S.; Chi, W.M.; Lee, C.H. A rapid and quantitative method to detect human circulating tumor cells in a preclinical animal model. BMC Cancer 2017, 17, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-L.; Tsai, M.-L.; Lin, C.-Y.; Hsu, K.-W.; Hsieh, W.-S.; Chi, W.-M.; Huang, L.-C.; Lee, C.-H. HDAC1 and HDAC2 Double Knockout Triggers Cell Apoptosis in Advanced Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 454. https://doi.org/10.3390/ijms20020454
Lin C-L, Tsai M-L, Lin C-Y, Hsu K-W, Hsieh W-S, Chi W-M, Huang L-C, Lee C-H. HDAC1 and HDAC2 Double Knockout Triggers Cell Apoptosis in Advanced Thyroid Cancer. International Journal of Molecular Sciences. 2019; 20(2):454. https://doi.org/10.3390/ijms20020454
Chicago/Turabian StyleLin, Ching-Ling, Ming-Lin Tsai, Chun-Yu Lin, Kai-Wen Hsu, Wen-Shyang Hsieh, Wei-Ming Chi, Li-Chi Huang, and Chia-Hwa Lee. 2019. "HDAC1 and HDAC2 Double Knockout Triggers Cell Apoptosis in Advanced Thyroid Cancer" International Journal of Molecular Sciences 20, no. 2: 454. https://doi.org/10.3390/ijms20020454