
BRD4 Inhibitor Inhibits Colorectal Cancer Growth and Metastasis

Abstract
:1. Introduction
2. Results and Discussion
2.1. BRD4 Is Highly Expressed in Colon Cancer Cells and Colon Cancer Tissues

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Characteristics | Number (%) of Patients (n = 45, Paired) | |
|---|---|---|
| Age (years) | 20–49 | 9 (20.0) |
| 50–64 | 20 (44.4) | |
| ≥65 | 16 (35.6) | |
| Gender | Male | 27 (60.0) |
| Female | 18 (40.0) | |
| Site of tumor | Ascending colon | 6 (13.3) |
| Transverse colon | 3 (6.7) | |
| Descending colon | 3 (6.7) | |
| Sigmoid colon | 13 (28.9) | |
| Rectum | 20 (44.4) | |
| Histologic differentiation | Well and moderately differentiated (≥50% gland formation) | 37 (82.2) |
| Poorly differentiated (<50% gland formation) | 7 (15.6) | |
| Missing | 1 (2.2) | |
| AJCC stage | I | 4 (8.9) |
| II-A | 6 (13.3) | |
| II-B | 11 (24.4) | |
| III-A | 0 (0.0) | |
| III-B | 13 (28.9) | |
| III-C | 5 (11.1) | |
| IV | 6 (13.3) | |
| T classification | T1 | 1 (2.2) |
| T2 | 3 (6.7) | |
| T3 | 12 (26.7) | |
| T4 | 29 (64.4) | |
| N classification | N0 | 21 (46.7) |
| N1 | 15 (33.3) | |
| N2 | 9 (20.0) | |
| M classification | M0 | 39 (86.7) |
| M1 | 6 (13.3) | |
| Relative fold change of cases expressing Brd4 (Tumor/Normal) | >1 | 31 (68.9) |
| <1 | 14 (31.1) | |

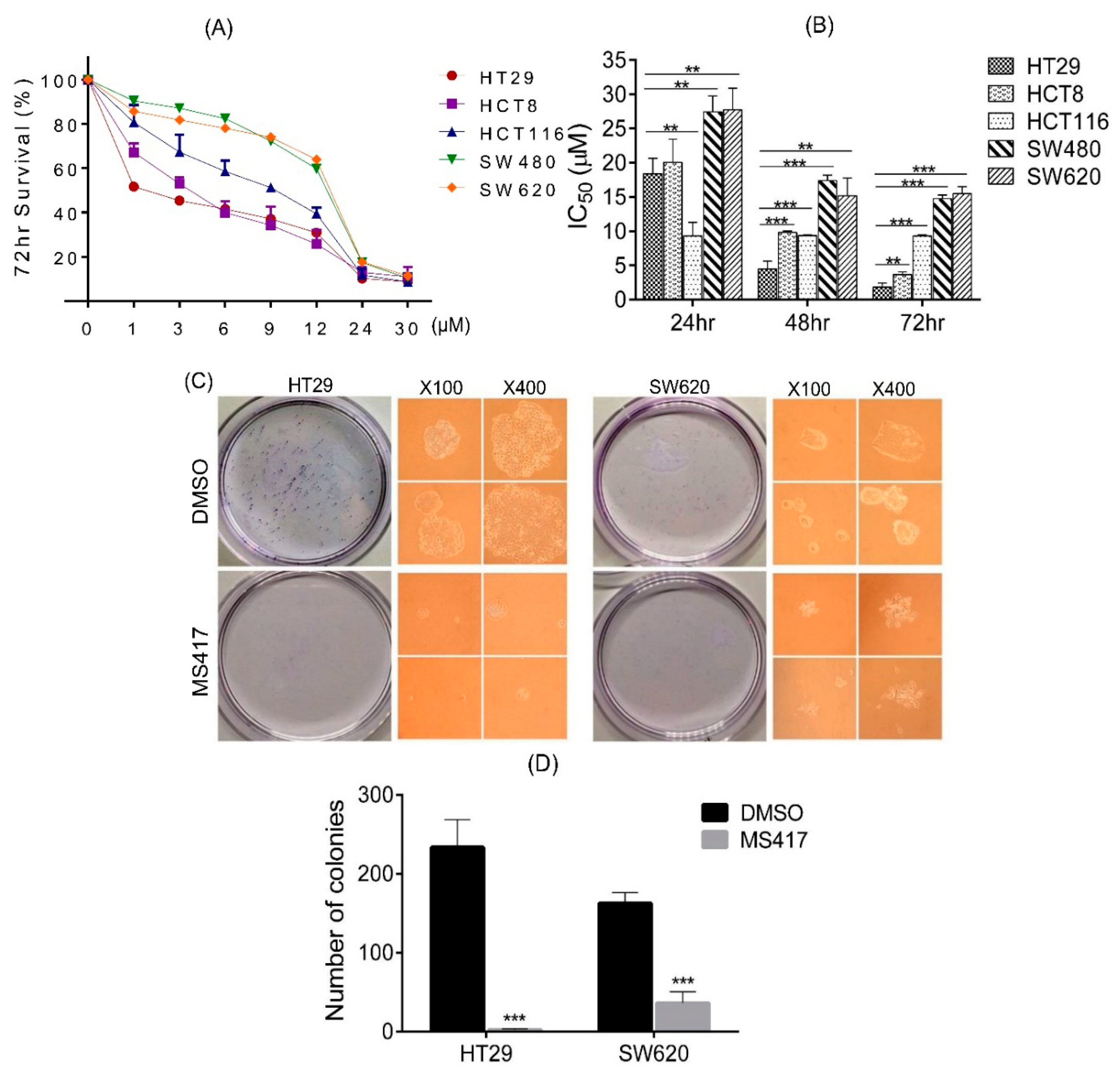
2.2. Colon Cancer Cell Proliferation Is Potently Inhibited by BRD4 Inhibitor in Vitro


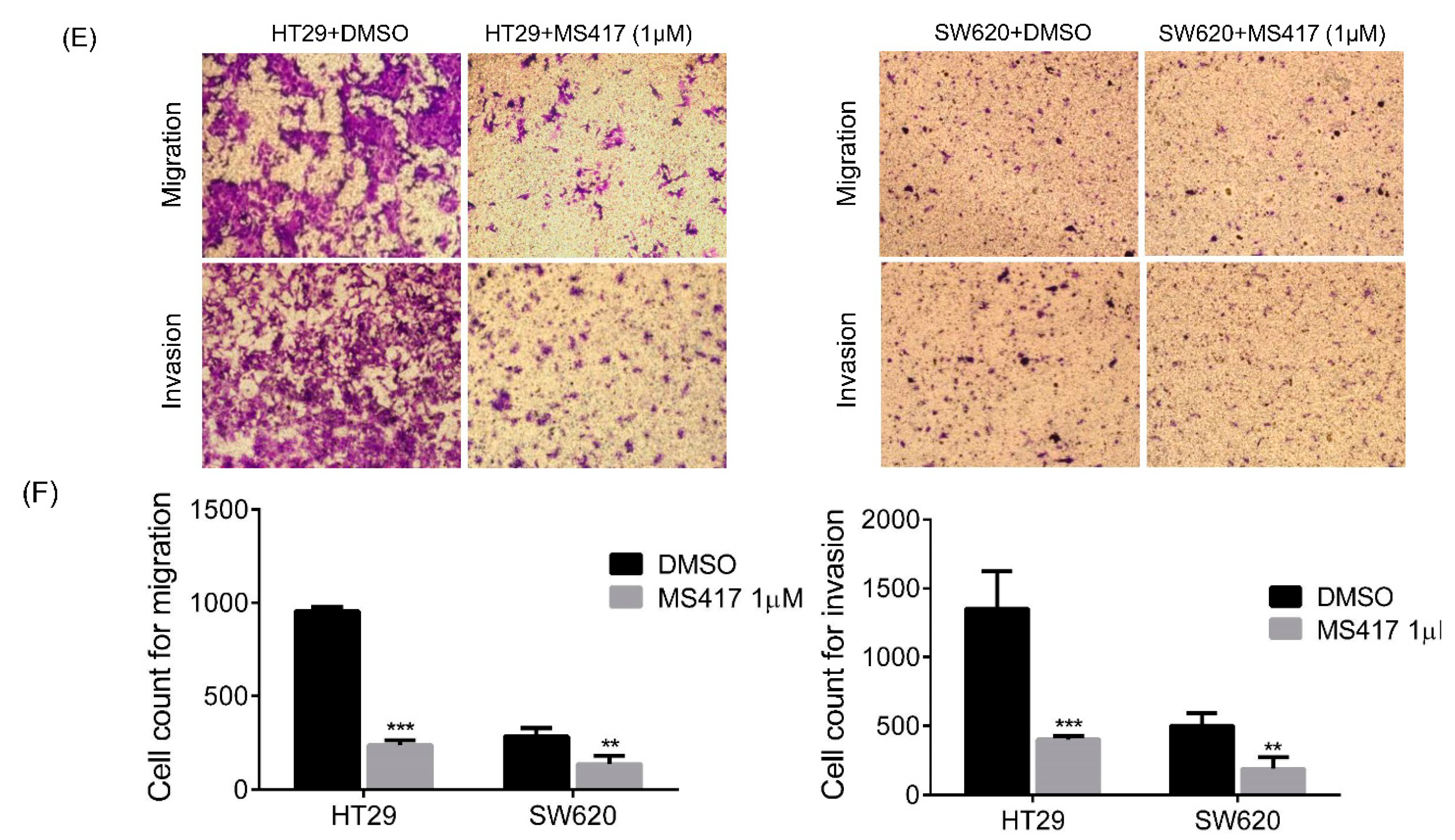
2.3. Colon Cancer Cell Migration and Invasion Are Reduced by BRD4 Inhibition in Vitro
2.4. BRD4 Inhibition Alters Protein Expression in CRC Cells


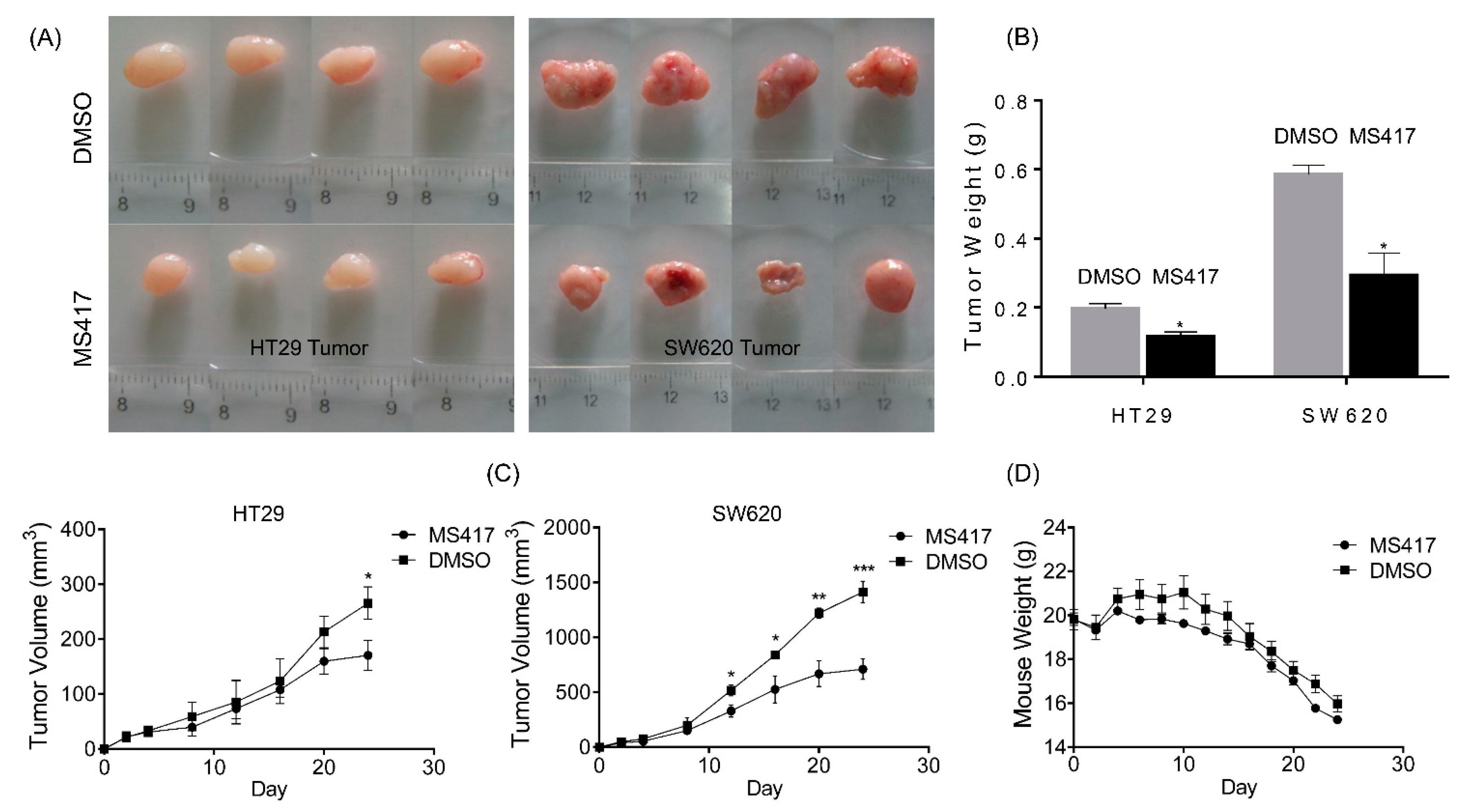
2.5. BRD4 Inhibition Impairs Colon Cancer Tumor Growth in Vivo

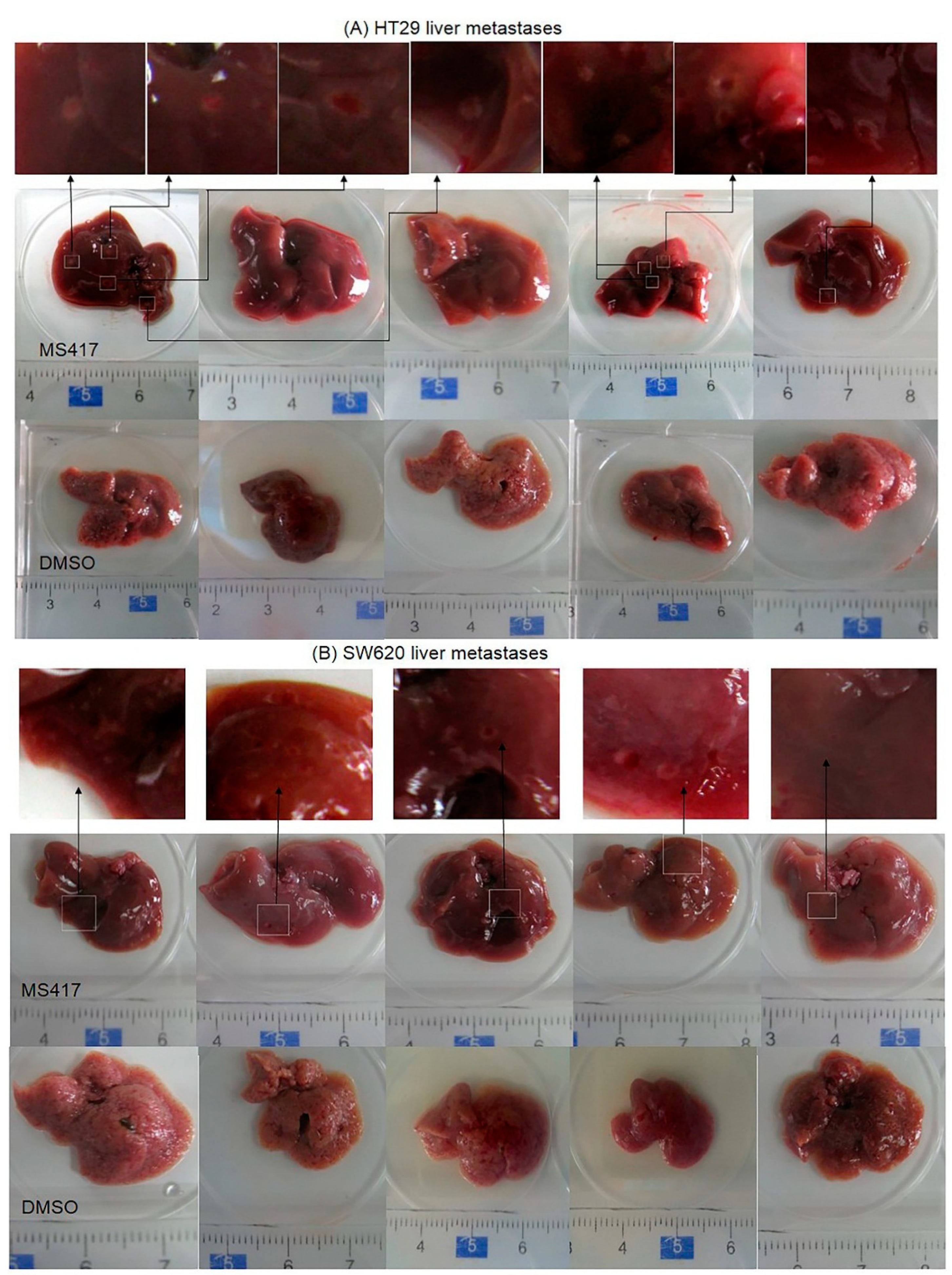
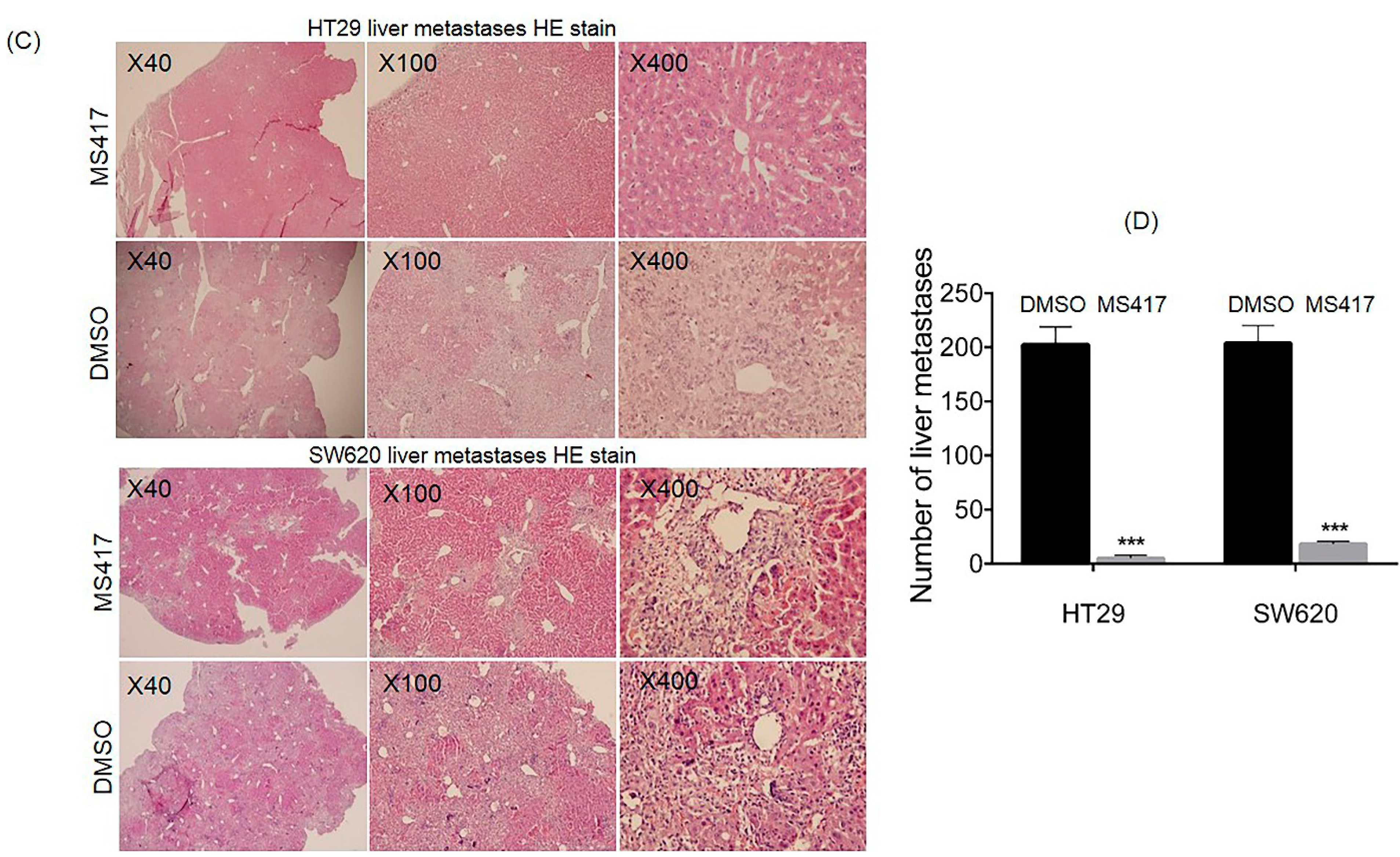
2.6. BRD4 Inhibition Suppressed Colon Cancer Liver Metastasis in Nude Mice


3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. Patient Samples
4.3. Real-Time PCR
4.4. MTT Assay
4.5. Colony Formation Assay
4.6. In Vitro Migration and Invasion Assay
4.7. Annexin V/PI Assays for Apoptosis
4.8. Western Blot Analysis
4.9. Mouse Xenograft and Liver Metastasis Model
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Desantis, C.; Jemal, A. Colorectal Cancer Statistics, 2014. CA Cancer J. Clin. 2014, 64, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Klajic, J.; Busato, F.; Edvardsen, H.; Touleimat, N.; Fleischer, T.; Bukholm, I.R.; Borresen-Dale, A.L.; Lonning, P.E.; Tost, J.; Kristensen, V.N. DNA Methylation Status of Key Cell Cycle Regulators such as CDKNA2/p16 and CCNA1 Correlates with Treatment Response to Doxorubicin and 5-Fluorouracil in Locally Advanced Breast Tumors. Clin. Cancer Res. 2014, 20, 6357–6366. [Google Scholar] [CrossRef] [PubMed]
- Alazzam, M.; Tidy, J.; Osborne, R.; Coleman, R.; Hancock, B.W.; Lawrie, T.A. Chemotherapy for Resistant or Recurrent Gestational Trophoblastic Neoplasia. Cochrane Database Syst. Rev. 2012, 12. [Google Scholar] [CrossRef]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone Acetylation in Chromatin Structure and Transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Van Engeland, M.; Derks, S.; Smits, K.M.; Meijer, G.A.; Herman, J.G. Colorectal Cancer Epigenetics: Complex Simplicity. J. Clin. Oncol. 2011, 29, 1382–1391. [Google Scholar]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and Ligand of a Histone Acetyltransferase Bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; Zeng, L.; Zhou, M.M. Structure and Acetyl-Lysine Recognition of the Bromodomain. Oncogene 2007, 26, 5521–5527. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K.; Nishiyama, A.; Jang, M.K.; Dey, A.; Ghosh, A.; Tamura, T.; Natsume, H.; Yao, H.; Ozato, K. The Bromodomain Protein BRD4 Stimulates G1 Gene Transcription and Promotes Progression to S Phase. J. Biol. Chem. 2008, 283, 9040–9048. [Google Scholar] [CrossRef] [PubMed]
- Lenburg, M.E.; Sinha, A.; Faller, D.V.; Denis, G.V. Tumor-Specific and Proliferation-Specific Gene Expression Typifies Murine Transgenic B Cell Lymphomagenesis. J. Biol. Chem. 2007, 282, 4803–4811. [Google Scholar] [CrossRef] [PubMed]
- French, C.A.; Miyoshi, I.; Kubonishi, I.; Grier, H.E.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4-NUT Fusion Oncogene: A Novel Mechanism in Aggressive Carcinoma. Cancer Res. 2003, 63, 304–307. [Google Scholar] [PubMed]
- Thompson-Wicking, K.; Francis, R.W.; Stirnweiss, A.; Ferrari, E.; Welch, M.D.; Baker, E.; Murch, A.R.; Gout, A.M.; Carter, K.W.; Charles, A.K.; et al. Novel BRD4-NUT Fusion Isoforms Increase the Pathogenic Complexity in NUT Midline Carcinoma. Oncogene 2013, 32, 4664–4674. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-Fusion Leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Ceribelli, M.; Kelly, P.N.; Shaffer, A.L.; Wright, G.W.; Xiao, W.; Yang, Y.; Mathews Griner, L.A.; Guha, R.; Shinn, P.; Keller, J.M.; et al. Blockade of Oncogenic IκB Kinase Activity in Diffuse Large B-Cell Lymphoma by Bromodomain and Extraterminal Domain Protein Inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 11365–11370. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC Dependence in Cancer by Inhibiting BET Bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Chen, Z.; Soucheray, M.; Carretero, J.; Kikuchi, E.; Tchaicha, J.H.; Gao, Y.; Cheng, K.A.; Cohoon, T.J.; Qi, J.; et al. Efficacy of BET Bromodomain Inhibition in Kras-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 6183–6192. [Google Scholar] [CrossRef] [PubMed]
- Crawford, N.P.; Alsarraj, J.; Lukes, L.; Walker, R.C.; Officewala, J.S.; Yang, H.H.; Lee, M.P.; Ozato, K.; Hunter, K.W. Bromodomain 4 Activation Predicts Breast Cancer Survival. Proc. Natl. Acad. Sci. USA 2008, 105, 6380–6385. [Google Scholar] [CrossRef] [PubMed]
- Alsarraj, J.; Walker, R.C.; Webster, J.D.; Geiger, T.R.; Crawford, N.P.; Simpson, R.M.; Ozato, K.; Hunter, K.W. Deletion of the Proline-Rich Region of the Murine Metastasis Susceptibility Gene Brd4 Promotes Epithelial-to-Mesenchymal Transition- and Stem Cell-Like Conversion. Cancer Res. 2011, 71, 3121–3131. [Google Scholar] [CrossRef] [PubMed]
- Alsarraj, J.; Faraji, F.; Geiger, T.R.; Mattaini, K.R.; Williams, M.; Wu, J.; Ha, N.H.; Merlino, T.; Walker, R.C.; Bosley, A.D.; et al. BRD4 Short Isoform Interacts with RRP1B, SIPA1 and Components of the LINC Complex at the Inner Face of the Nuclear Membrane. PLoS One 2013, 8, e80746. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.F.; Fontanals-Cirera, B.; Gaziel-Sovran, A.; Guijarro, M.V.; Hanniford, D.; Zhang, G.; Gonzalez-Gomez, P.; Morante, M.; Jubierre, L.; Zhang, W.; et al. BRD4 Sustains Melanoma Proliferation and Represents a New Target for Epigenetic Therapy. Cancer Res. 2013, 73, 6264–6276. [Google Scholar] [CrossRef] [PubMed]
- Floyd, S.R.; Pacold, M.E.; Huang, Q.; Clarke, S.M.; Lam, F.C.; Cannell, I.G.; Bryson, B.D.; Rameseder, J.; Lee, M.J.; Blake, E.J.; et al. The Bromodomain Protein Brd4 Insulates Chromatin from DNA Damage Signalling. Nature 2013, 498, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Kumar, K.; Knab, L.M.; Chow, C.R.; Raza, S.S.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. BET Bromodomain Inhibitors Block Growth of Pancreatic Cancer Cells in Three-Dimensional Collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.M.; Huidobro, C.; Urdinguio, R.G.; Mangas, C.; Soldevilla, B.; Dominguez, G.; Bonilla, F.; Fernandez, A.F.; Fraga, M.F. Aberrant Epigenetic Regulation of Bromodomain BRD4 in Human Colon Cancer. J. Mol. Med. 2012, 90, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, R.; Zhong, Y.; Plotnikov, A.N.; Zhang, W.; Zeng, L.; Rusinova, E.; Gerona-Nevarro, G.; Moshkina, N.; Joshua, J.; et al. Down-Regulation of NF-κB Transcriptional Activity in HIV-Associated Kidney Disease by BRD4 Inhibition. J. Biol. Chem. 2012, 287, 28840–28851. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET Bromodomain-Targeting Compounds Reactivate HIV from Latency Via a Tat-Independent Mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhong, Y.; Li, X.; Chen, H.; Jim, B.; Zhou, M.M.; Chuang, P.Y.; He, J.C. Role of Transcription Factor Acetylation in Diabetic Kidney Disease. Diabetes 2014, 63, 2440–2453. [Google Scholar] [CrossRef] [PubMed]
- Lavilla-Alonso, S.; Abo-Ramadan, U.; Halavaara, J.; Escutenaire, S.; Tatlisumak, T.; Saksela, K.; Kanerva, A.; Hemminki, A.; Pesonen, S. Optimized Mouse Model for the Imaging of Tumor Metastasis upon Experimental Therapy. PLoS One 2011, 6, e26810. [Google Scholar] [CrossRef] [PubMed]
- Maamer-Azzabi, A.; Ndozangue-Touriguine, O.; Breard, J. Metastatic SW620 Colon Cancer Cells are Primed for Death when Detached and can be Sensitized to Anoikis by the BH3-Mimetic ABT-737. Cell. Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chiang, C.M. The Double Bromodomain-Containing Chromatin Adaptor Brd4 and Transcriptional Regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; He, N.; Zhou, Q. Brd4 Recruits P-TEFb to Chromosomes at Late Mitosis to Promote G1 Gene Expression and Cell Cycle Progression. Mol. Cell. Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 Activates P-TEFb for RNA Polymerase II CTD Phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Dey, A.; Miyazaki, J.; Ozato, K. Brd4 is Required for Recovery from Antimicrotubule Drug-Induced Mitotic Arrest: Preservation of Acetylated Chromatin. Mol. Biol. Cell 2006, 17, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.S.; Zhang, B.; Smith, J.J.; Jayagopal, A.; Barrett, C.W.; Pino, C.; Russ, P.; Presley, S.H.; Peng, D.; Rosenblatt, D.O.; et al. BVES Regulates EMT in Human Corneal and Colon Cancer Cells and Is Silenced via Promoter Methylation in Human Colorectal Carcinoma. J. Clin. Investig. 2011, 121, 4056–4069. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the Interaction of BRD4 with Diacetylated Twist Suppresses Tumorigenesis in Basal-Like Breast Cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail Genes as Inducers of Cell Movement and Survival: Implications in Development and Cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-Activator ZEB1 Promotes Tumorigenicity by Repressing Stemness-Inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Fotoohi, K.; Jansen, G.; Assaraf, Y.G.; Rothem, L.; Stark, M.; Kathmann, I.; Gregorczyk, J.; Peters, G.J.; Albertioni, F. Disparate Mechanisms of Antifolate Resistance Provoked by Methotrexate and its Metabolite 7-Hydroxymethotrexate in Leukemia Cells: Implications for Efficacy of Methotrexate Therapy. Blood 2004, 104, 4194–4201. [Google Scholar] [CrossRef] [PubMed]
- Yurgel, V.C.; Oliveira, C.P.; Begnini, K.R.; Schultze, E.; Thurow, H.S.; Leon, P.M.; Dellagostin, O.A.; Campos, V.F.; Beck, R.C.; Guterres, S.S.; et al. Methotrexate Diethyl Ester-Loaded Lipid-Core Nanocapsules in Aqueous Solution Increased Antineoplastic Effects in Resistant Breast Cancer Cell Line. Int. J. Nanomed. 2014, 9, 1583–1591. [Google Scholar]
- Wang, T.P.; Hsu, S.H.; Feng, H.C.; Huang, R.F. Folate Deprivation Enhances Invasiveness of Human Colon Cancer Cells Mediated by Activation of Sonic Hedgehog Signaling through Promoter Hypomethylation and Cross Action with Transcription Nuclear Factor-κB Pathway. Carcinogenesis 2012, 33, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.I.; Ahn, J.H.; Lee, K.T.; Shih, I.; Choi, J.H. RSF1 is a Positive Regulator of NF-κB-Induced Gene Expression Required for Ovarian Cancer Chemoresistance. Cancer Res. 2014, 74, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Zhou, J.; Ye, F.; Xiong, H.; Peng, L.; Zheng, Z.; Xu, F.; Cui, M.; Wei, C.; Wang, X.; et al. BRD4 Inhibitor Inhibits Colorectal Cancer Growth and Metastasis. Int. J. Mol. Sci. 2015, 16, 1928-1948. https://doi.org/10.3390/ijms16011928
Hu Y, Zhou J, Ye F, Xiong H, Peng L, Zheng Z, Xu F, Cui M, Wei C, Wang X, et al. BRD4 Inhibitor Inhibits Colorectal Cancer Growth and Metastasis. International Journal of Molecular Sciences. 2015; 16(1):1928-1948. https://doi.org/10.3390/ijms16011928
Chicago/Turabian StyleHu, Yuan, Jieqiong Zhou, Fei Ye, Huabao Xiong, Liang Peng, Zihan Zheng, Feihong Xu, Miao Cui, Chengguo Wei, Xinying Wang, and et al. 2015. "BRD4 Inhibitor Inhibits Colorectal Cancer Growth and Metastasis" International Journal of Molecular Sciences 16, no. 1: 1928-1948. https://doi.org/10.3390/ijms16011928